
Wei Yang
Key Laboratory of Marine Food Quality and Hazard Controlling Technology, College of Life Science, Hangzhou, Zhejiang Province, China
Xianjun Dai
Key Laboratory of Marine Food Quality and Hazard Controlling Technology, College of Life Science, Hangzhou, Zhejiang Province, China
Mingqi Liu
Key Laboratory of Marine Food Quality and Hazard Controlling Technology, College of Life Science, Hangzhou, Zhejiang Province, China
American Journal of Food Technology
Year: 2016 | Volume: 11 | Issue: 3 | Page No.: 100-107







ABSTRACT
Listeria monocytogenes is an emerging bacterial food borne pathogen, which could survive common stress levels and often cause listeriosis. To control its infection, it is necessary to establish a sensitive, specific, rapid technology for detecting Listeria monocytogenes. In this study, invasion-associated-protein (IAP) was used as a target gene. After prokaryotic cloning and expression of the gene, the specific membrane protein P60 of Listeria moncytogenes was obtained. The P60 was injected into New Zealand white rabbits to get the antibody. The prepared antibody was conjugated with magnetic-bead to get immunomagnetic-beads. Enzyme-linked immunosorbent assays method based on immunomagetic-bead (IMB-ELISA) would be developed through immunomagetic-bead (IMB) collecting Listeria moncytogenes. The results showed that the sensitivity of the IMB-ELISA was 1.2x106 CFU mL–1, the limit of detection was 31 CFU mL–1. The IMB-ELISA is a quick and accurate method for the detection of Listeria moncytogenes.
PDF Abstract XML References Citation
Received: June 11, 2015;
Accepted: February 24, 2016;
Published: April 15, 2016
How to cite this article
Wei Yang, Xianjun Dai and Mingqi Liu, 2016. Development of an Indirect Competitive Enzyme-linked Immunosorbent Assays Method Based on Immunomagetic-bead for Analyzing Listeria monocytogenes in Food. American Journal of Food Technology, 11: 100-107.
DOI: 10.3923/ajft.2016.100.107
URL: https://scialert.net/abstract/?doi=ajft.2016.100.107
DOI: 10.3923/ajft.2016.100.107
URL: https://scialert.net/abstract/?doi=ajft.2016.100.107
INTRODUCTION
Listeria monocytogenes (L. monocytogenes) is an emerging bacterial foodborne pathogen responsible for listeriosis, an illness characterized by meningitis, encephalitis and septicaemia. Infection could result in cutaneous lesions and flu-like symptoms (Guo et al., 2011; Churchill et al., 2006). In pregnant women, the pathogen could cause bacteraemia and stillbirth or premature birth of the fetus and the mortality rate for those contracting listeriosis is approximately 20% (Yan et al., 2006). Listeria monocytogenes has a complex pathogenicity mechanism with several virulence factors which improve transmission by direct cell-to-cell infection.
As one of four major food borne pathogens by World Health Organization (WHO) (Buzby and Roberts, 1997), L. monocytogenes could become one of the important risk factors in food and it is essential to detect the pathogen of low numbers in food samples. Traditional testing methods for L. monocytogenes in food include the isolation of the suspected colonies and examination of biochemical reaction, hemolysis and typical motion. For these above methods, selective enrichment culture is the indispensable step, which means the long time, high cost and low sensitivity (Churchill et al., 2006).
In view that the traditional detection is time-consuming in enrichment culture, it is necessary to establish a sensitive, specific and rapid technology for detecting L. monocytogenes in food to improve sensitivity and detecting efficiency, several new techniques had been developed based on immunology or molecular biology.
The enzyme-linked immunosorbent assays (ELISA) are widely used as screening method for detecting chemical residue and microorganism in foods and environmental samples under the restricted levels (Wanatabe et al., 2001). The ELISA has several advantages of rapidness with short incubation time and simplicity without complicated clean-up procedures in analysis. The ELISA has been extensively used for screening of bacteria including Escherichia coli, Staphylococcus aureus and other bacteria in food due to its sensitivity, specificity and rapidity and simplicity (He et al., 2004).
Immunomagnetic separation (immunomagentic bead-based separation, IMS) is one of the separation technology methods based on the specific antigen-antibody reaction. Immunomagnetic separation makes use of antibody specificity towards a pathogen to concentrate that pathogen before other methods are used to amplify and identify the bacteria. Antibodies are attached to beads and added to a homogenized sample. Any pathogen with affinity for the antibody should attach to the immunomagentic beads (IMB) complex. When the sample is mixed with the IMB, the pathogen can be wrapped in the beads because of the specifically reaction between the pathogen and antibody which is conjugated to the Magentic Beads (MB). The compounds can be rapidly separated by NdFeB magnet (Fitzmaurice et al., 2004; Li and Wu, 2008). Therefore, this technology not only has the advantages of solid phase immunological reactions, but also can reduce the detection time and improve the detecting sensitivity (Wang et al., 2014). With these advantages, IMS is widely used in biomedical and food safety, such as the cell separation and the pathogen detection.
In this study, P60 protein of L. monocytogenes was expressed as antigen to prepare the antibody, because P60 protein has hydrolase and amidase activity and can stimulate B-lymphocyte and T-lymphocyte to produce the immune responses, it is a virulence factor of L. monocytogenes (Wang et al., 2006a; Bubert et al., 1992; Lu et al., 2010; Zhu and Ye, 2007). Magnetic-bead would be conjugated with polyclonal antibody to get immunomagnetic-beads, enzyme-linked immunosorbent assays method based on immunomagetic-bead (IMB-ELISA) was established through IMB collecting L. monocytogenes.
MATERIALS AND METHODS
Animal preparation: Two New Zealand white rabbits were obtained from the experimental Animal Research Institute of Hangzhou Normal University. Animal welfare and experimental procedures were carried out in accordance with the guide for the care and use of laboratory animals were approved by the animal ethics committee of Hangzhou Normal University, Zhejiang, China.
Bacterial strains: Strains of Listeria monocytogenes, Escherichia coli O157 and Enterococcus faecalis were preserved strains in this laboratory; E. coli DH5α and E. coli BL21 (DE3) were purchased from CW biotech (Beijing, China).
Reagents: Premix polymerase, pMD-18T, DNA Ligation Kit, Quick Cut BamHI and Quick Cut XhoI were purchased from TAKRA (Japan), Freund's Complete Adjuvant (FCA) and Freund's Incomplete Adjuvant (FIA) were purchased from Sigma (USA). Goat anti-rabbit IgG/HRP, nucleic acid stain, protein molecular weight marker, DNA marker, the SanPrep column DNA gel extraction kit, PBS and TBS were purchased from Sangon Biotech (Shanghai, China), the plasmid Mini Kit was purchased from OMEGA (USA), TMB-component color liquid was purchased from Aladdin (Shanghai, China), immunomagentic beads were purchased from Huier biotech (Luoyang, China), PVDF membrane, ponceau S staining solution, developing fixing kit and ECL chemiluminescence were purchased from beyotime Biotechnology (Jiangsu, China), other reagents were routine laboratory reagents, analytical grade, they were purchased from Mike Chemical Instrument Corporation Limited (Hangzhou, China).
Cloning IAP gene: Genome DNA of L. monocytogenes was extracted from overnight cultures and was used as template for PCR amplification of the gene encoding protein P60. The cloning primers were designed as follows: 5'-CGGGATCCATGAATATGAAAAAAGCAAC-3' (forward primer, containing BamHI digestion site); 5'-CCCTCGAGTTATACGC GACCGAAGCCAA-3’ (reverse primer, containing XhoI digestion site). The PCR amplified products were purified by 1% Agarose Gel Electrophoresis (AGE) and inserted into pMD-18T simple vector. The obtained pMD18T-IAP vector was transformed into competent E. coli DH5α and sequenced to confirm nucleotide identity. The correct pMD18T-IAP plasmid and pET-30a plasmid DNA were digested with BamHI and Xho I overnight at 37°C. The gene was directly ligated into the vector with rapid DNA ligation Kit and the recombinant plasmid pET30a-IAP was transformed into E. coli BL21 (DE3).
Expression and identification of P60 gene in E. coli: Single clone of E. coli BL21 (DE3) harboring the pET30a-IAP vector was inoculated into 5 mL LB medium containing 50 μg mL–1 kanamycin and was cultured at 37°C and 150 rpm on a rotary shaker overnight. One milliliter of these cells was inoculated into 50 mL 2×YT medium. The cells were incubated at 37°C for 2 h on a rotary shaker until the cell concentration of OD600 = 0.5-0.7 and IPTG (Isopropylthiogalactoside) was added to induce protein expression by a final concentration of 1 mM. After induction for 8 h at 28°C, cells were harvested by centrifugation at 8500 rpm for 10 min at 4°C. The precipitate was resuspended with PBS (pH 7.4) and the expression protein can be obtained after the sonication. Proteins were detected directly by 12% SDS-PAGE and the protein bands were stained by coomassie brilliant blue.
For Western blot analysis, electroblotting of proteins on PVDF membrane was carried out using wet method in a transfer buffer (25 mM tris, 192 mM glycine and 20% methanol) at 90 mA for 1 h. After blocking the membrane with 5% skim milk in TBS overnight at 4°C, the blot was probed with rabbit antibody L. monocytogenes polyclonal antibody as the primary antibody in TBS for 1 h at room temperature by shaking, washed three times (TBS with 0.1% tween20) and incubated with goat anti-rabbit IgG-HRP as the secondary antibody in TBS for 60 min at room temperature by shaking. The ECL chemiluminescence was used to detect the target band.
Immunization for rabbits and antiserum preparation: Two New Zealand white rabbits were immunized with intramuscular injections of the purified protein P60 for three times. After the third booster, each rabbit was bled from the ear vein on the 7th day and the serum was separated through centrifuge. The positive serum of above 1:12800 titers was used in the following experiment. Antibody was purified from separated serum by bitterness-ammonium sulfate precipitation.
Combination of magnetic bead and the antibody: Activated beads was been washed with 1 mL MES (pH 6.0) buffer for 2 times. Five hundred microlitter of the antibody proteins were added into the solution of activated beads for overnight at room temperature. The mixed solution was placed on a magnetic separation rack to obtain the magnetic beads-antibody conjugate (IMB). The IMB was washed with 1 mL MES (pH 6.0) buffer for 2 times and resuspended by 1 mL of PBS, which included 0.1% Bovine Serum Albumin (BSA) for storage at 4°C.
Establishment of the standard curve and the sensitivity determination: The antibody (antiserum or IMB) was diluted with PBS, antigen-coated was diluted with CBS, L. moncytogenes of 103, 104, 105, 106, 107, 108, 109 and 1010 CFU mL–1 were used as the coated antigen to do the phalanx titration. The antibodies and antigen dilutions were selected as the optimal working concentration, when the OD450 was approximately equal to 1. The optimal working concentration of antigen and antibody (antiserum or IMB) were used to establish the standard curve. The ELISA plates were coated with 100 μL 105 CFU mL–1 cultures in each well and the 90 μL antibody were mixed with 10 μL antigen of final concentration 0, 102, 103, 104, 105, 106, 107 and 108 CFU mL–1. The specificity was determined with the IC-ELISA and the half maximal Inhibitory Concentration (IC50, the concentration causing 50% inhibition of binding) and the detectable limitation (IC10, the concentration causing 90% inhibition of binding) were calculated.
In the result:
where, B is the absorbance of well with the competitor and B0 is the absorbance of well without the competitor.
Specific detection: The assay specificity was evaluated by testing the cross-reactivity (CR) of the antibody with other pathogenic microorganisms, Escherichia coli O157 and Enterococcus faecalis. The CR values were calculated according to the following:
Repeatability detection
Repeatability tests with the same plate: According to the established ELISA methods, the antiserum and the immunomagnetic beads-antibody conjugate was respectively used as the antibody to determine the L. monocytogenes in samples of known concentrations. The colonies concentration was 105, 106 and 107 CFU mL–1, respectively and each concentration was repeated in 10 different wells. The antibody was replaced by PBS in the blank well. Coefficient of variation of the same plate was calculated under the different operating methods and different colonies.
Repeatability tests between different plates: According to the established ELISA methods, the anti-serum and the immunomagnetic beads-antibody conjugate was, respectively used as the antibody to determine the L. monocytogenes in samples. The same concentrations of L. monocytogenes were determined in three microtiter plates, respectively and each concentration was repeated in 10 different wells in one plate. The antibody was replaced by PBS in the blank well. Coefficient of variation of the same plate was calculated according to the different operating methods and different plates.
Recovery study in samples: The different concentrations of L. monocytogenes were used as samples in two established methods. In the IC-ELISA, each 0.1 mL sample was coupled with 0.9 mL antiserum; in the IMB-ELISA, each 0.1 mL sample was coupled with 0.9 mL immunomagnetic beads-antibody. The mixture was shocked in the sample mixing device for 45 min at room temperature and separated by NdFeB magnet for 3 min in order to capture the L. monocytogenes in samples.
RESULTS
Listeria monocytogenes is a serious threat to the food industry as a psychrotrophic species and could survive the most common stress levels presented in the food processing environment, such as high salinity, acidity, refrigeration temperatures and low water activity. In this study, the IMB-ELISA method was developed for the detection of the L. monocytogenes using the polyclonal antibody raised against P60 protein.
The cloning of IAP: The length of amplified product was between 1000 and 1500 bp (Fig. 1) and the sequence of IAP gene was 1437 bp. The BLAST result demonstrated the nucleotide sequence of the cloned DNA shared 99% identity with IAP gene in GenBank and there was no difference of amino acid sequence between them.
Expression and identification of P60 in E. coli: After double digestion with BamHI and XhoI, pET30a and IAP gene were combined into the recombinant vector, which was transformed it into competent E. coli BL21 (DE3). The PCR analysis was applied to check the proper insertion of the gene in the recombinant plasmid using T7 promoter-specific primers. The protein expression was monitored by SDS-PAGE and Western-blot analysis (Fig. 2).
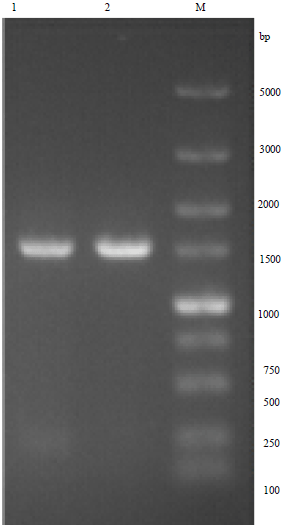 |
| Fig. 1: | PCR amplification product of IAP gene, Lanes 1 and 2: PCR identify of IAP gene, Lane M: DNA marker |
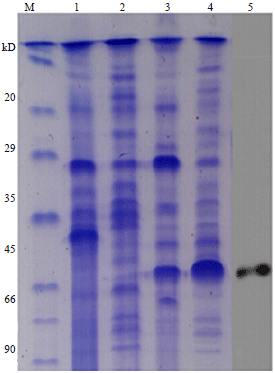 |
| Fig. 2: | Analysis of expressed proteins by gradient 12% SDS-PAGE and Western blot: Lane M, protein marker, Lanes 1 and 2: Sonication precipitate and the supernatant fraction of cell lysate with empty vector pET30a, Lane 3: Supernatant fraction of cell lysate, Lane 4: Sonication precipitate of cell lysate, where specific protein band could be found and Lane 5: Western blotting analysis of P60 with polyclonal antibody against L. monocytogenes culture as primary antibody and goat anti-rabbit IgG-HRP as secondary antibody |
There was a clear band with a molecular weight of approximately 60 kD in the lane 4, which was different from lane 2, so we inferred this band maybe the P60, the Western blotting was done with polyclonal antibody against L. monocytogenes culture used as primary antibody and goat anti-rabbit IgG-HRP used as secondary antibody, the result was shown in lane 5, it can obtain a specific band, which has a same size with lane 4. It illustrated the protein P60 was successfully expressed.
Determination of the optimal working concentrations: The antibodies and antigen dilutions were selected as the optimal working concentration, when he OD450 was approximately equal to 1. In the IC-ELISA, the OD450 value was 0.982, the concentration of antigen was 106 CFU mL–1 and the diluted antibody against P60 protein was 1:800. The above step could be repeated for IMB-ELISA examination, in which antibody was replaced by IMB.
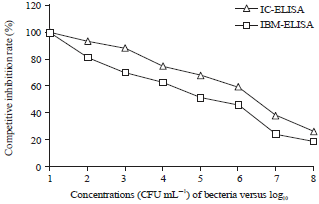 |
| Fig. 3: | Standard curves for two different ELISA methods |
It was determined that the optimal concentration of antigen was 105 CFU mL–1, when OD450 value was 1.042 and the optimal rate of diluted antibody was 1:800 for IMB-ELISA examination.
Establishment of the standard curve and the sensitivity determination: As is shown in Fig. 3, the calculated curve was y = -10.482x+115.91, R2 = 0.9667 in the IC-ELISA and y = -11.094x+106.91, R2 = 0.9812 in the IMB-ELISA. The IC50 values were 2×107 and 1.2×106 CFU mL–1 and IC10 values were 316 and 31 CFU mL–1, respectively for IC-ELISA and IMB-ELISA. The results indicated the established IMB-ELISA was more sensitive than IC-ELISA for monitoring L. monocytogenes in food.
Specific detection: The specificity was respectively examined for the IC-ELISA and IMB-ELISA through detecting Escherichia coli O157 and Enterococcus faecalis. The results showed that CR values were less than 0.1% for the above-mentioned pathogenic microorganisms, which suggested the prepared antibody possessed a high specificity for detection of L. monocytogenes in samples.
Repeatability detection
Detection of the repeatability with the same plate: According to the determination of L. monocytogenes samples of 105, 106 and 107 CFU mL–1, the coefficient of variation were, respectively 6.87, 4.77 and 2.61 for the IC-ELISA and 4.28, 3.16 and 1.90% for the IMB-ELISA in the same plate. The coefficient of variation of less than 10% indicated two detection methods had a good repeated accuracy and the IMB-ELISA had a better repeatability in the same plate (Table 1).
Detection of the repeatability between different plates: Listeria monocytogenes of 105 CFU mL–1 was determined in 3 μL plates, respectively. For the IC-ELISA, the coefficient of variation was 6.36 between three different plates and the coefficient of variation was 2.69 for the IMB-ELISA.
| Table 1: | Result of the repeatability test in one plate |
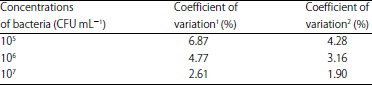 | |
1: IC-ELISA, 2: IMB-ELISA, IC-ELISA: Inhibitory concentration-enzyme linked immuno sorbent assay and IMB-ELISA: Immunomagnetic bead-enzyme linked immunosorbent assay | |
| Table 2: | Result of the repeatability test in three plates |
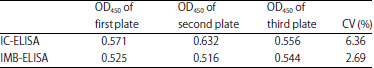 | |
| Table 3: | Recovery study of Listeria moncytogenes in samples |
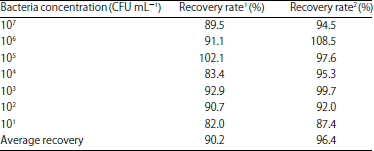 | |
| 1: IC-ELISA, 2: IMB-ELISA | |
The coefficient of variation of less than 10% indicated two detection methods have a good repeatability and the IMB-ELISA has better repeatability between different plates (Table 2).
Recovery study in samples: According to these established two detection methods (IC-ELISA and the IMB-ELISA), the recovery rates were shown in Table 3. When the concentration of L. monocytogenes was more than 101 CFU mL–1, the average recovery rate was 90.2% in the IC-ELISA and 96.4% in the IMB-ELISA, respectively.
DISCUSSION
Listeria monocytogenes is a serious threat to the food industry as a psychrotrophic species and could survive the most common stress levels presented in the food processing environment, such as high salinity, acidity, refrigeration temperatures and low water activity. In view that the traditional detection is time-consuming in enrichment culture, several new techniques had been developed based on immunology or molecular biology.
After analysis of the virulence genes of L. monocytogenes, conventional PCR and the real-time PCR were used in the specific detection (Wang et al., 2006b; Xu et al., 2007) and FRET-PCR (fluorescence resonance energy transfer based PCR), oligonucleotide probe detection and DNA chip technology were developed in the recent years (Moreno et al., 2011; Suo et al., 2010). A combination of PCR and DNA probe was prepared for recognizing a specific region of the internalin gene, which is responsible for the production of one of by most important pathogenic factors of L. monocytogenes by Ingianni et al. (2001). Five individual oligoprobes were selected for identification of each Listeria species by Sergeev et al. (2004), in which Cy5-labeled iap DNA from L. monocytogenes strain LM82 was hybridized specifically to the oligoprobes corresponding to the L. monocytogenes iap gene. It has been shown that fluorescence resonance energy transfer (FRET)-based PCR was used to detect the L. monocytogenes, including the TaqMan assay and molecular beacons (Koo and Jaykus, 2003). The limits of these above detection methods were 60-103 CFU mL–1 and the sensitivities were 28-60 CFU mL–1 based on molecular biology methods for the detection of L. monocytogenes (Ingianni et al., 2001; Koo and Jaykus, 2003; Sergeev et al., 2004; Wang et al., 2006b; Xu et al., 2007; Moreno et al., 2011; Suo et al., 2010).
Based on the immune detection, Luo et al. (2009) gold immune-chromatography assay coupled with monoclonal antibodies against L. monocytogenes. The double-antibody sandwich indirect ELISA method was, respectively developed for detection of L. monocytogenes (Chen, 2010; Duan et al., 2010). The bead array and sandwich ELISA was developed for detection of L. monocytogenes by Karoonuthaisiri et al. (2015). In these ELISA study, the limit of detection for L. monocytogenes was 1.7×105-3.3×106 CFU mL–1, but the sensitivities were not mentioned in these references (Chen, 2010; Duan et al., 2010; Karoonuthaisiri et al., 2015).
CONCLUSION
In this study, the IMB-ELISA method was developed for the detection of the L. monocytogenes using the polyclonal antibody raised against P60 protein. The sensitivity was 1.2×106 CFU mL–1 and the limit of detection was 31 CFU mL–1. There was lower detection limit and higher sensitivity than that of PCR methods. However, these PCR detection methods are need to prepare the template from a higher concentration of bacteria culture and more time was needed to improve the bacterial colony in the sample. Compared with the PCR related methods, IMB-ELISA had a lower detection limit and higher sensitivity. However, these PCR detection methods are need to prepare the template from a higher concentration of bacteria culture and more time was needed to improve the bacterial colony in the sample. To the many of these reported ELISA methods, the detection limit of IMB-ELISA was lower and the sensitivity was higher. In consideration that IMB can quick enrich the target bacteria and the target compounds can be rapidly separated by NdFeB magnet, IMB-ELISA had a better advantage than the reported detecting method of ELISA. On the other hand, the detection method of IMB-ELISA had a better recovery and repeatability, which could basically meet the needs of the detection of the L. monocytogenes.
The results of this study demonstrate that the IMB-ELISA using the polyclonal antibody raised against P60 protein is a convenient and sensitive method and could be used for the rapid detection of L. monocytogenes in the future.
ACKNOWLEDGMENT
This research was financially supported by funding (2012C03009-2) from the Science Technology Department of Zhejiang Province, China.
REFERENCES
- Bubert, A., M. Kuhn, W. Goebel and S. Kohler, 1992. Structural and functional properties of the p60 proteins from different Listeria species. J. Bacteriol., 174: 8166-8171.
Direct Link - Buzby, J.C. and T. Roberts, 1997. Economic costs and trade impacts of microbial foodborne illness. World Health Stat. Q., 50: 57-66.
PubMed - Churchill, R.L.T., H. Lee and J.C. Hall, 2006. Detection of Listeria monocytogenes and the toxin listeriolysin O in food. J. Microbiol. Methods, 64: 141-170.
CrossRefDirect Link - Duan, X., X. Huang, L.F. Huang, H. Wei and W.H. Lai, 2010. [Detection of Listeria monocytogenes in food by sandwich ELISA]. Food Sci., 31: 272-276, (In Chinese).
Direct Link - Fitzmaurice, J., G. Duffy, B. Kilbride, J.J. Sheridan, C. Carroll and M. Maher, 2004. Comparison of a membrane surface adhesion recovery method with an IMS method for use in a polymerase chain reaction method to detect Escherichia coli O157:H7 in minced beef. J. Microbiol. Methods, 59: 243-252.
CrossRefDirect Link - Guo, G.P., H.M. Ge, Y. Wang, W.G. Zhang and P. Ni, 2011. Research advancement of detection methods of Listeria monocytogenes. Food Nutr. China, 17: 12-15.
Direct Link - He, Q.Q., J.B. Yang, C.F. Xiao and T.C. Zuo, 2004. Development of double-antibodies-sandwich-ELISA method for detecting HSP70 in plasma and its primary application. J. Environ. Occup. Med., 21: 27-30.
Direct Link - Ingianni, A., M. Floris, P. Palomba, M.A. Madeddu, M. Quartuccio and R. Pompei, 2001. Rapid detection of Listeria monocytogenes in foods, by a combination of PCR and DNA probe. Mol. Cell. Probes, 15: 275-280.
CrossRefDirect Link - Koo, K. and L.A. Jaykus, 2003. Detection of Listeria monocytogenes from a model food by fluorescence resonance energy transfer-based PCR with an asymmetric fluorogenic probe set. Applied Environ. Microbiol., 69: 1082-1088.
CrossRefDirect Link - Li, Y.J. and S.Q. Wu, 2008. Application of immunomagnetic separation technique in detection of food-borne diseases. Sci. Technol. Food Ind., 12: 248-251.
Direct Link - Lu, T., H.T. Wu, Z.M. Cao and X.H. Wang, 2010. [Cloning and expression of iap gene from Listeria monocytogenes and purification of protein p60]. Food Sci., 31: 157-161, (In Chinese).
Direct Link - Luo, Y., Z. Jia, X. Ding, L. Fang, Z. Zheng and C. He, 2009. Gold immunochromato-graphy assay coupled with monoclonal antibodies against Listeria monocytogenes for detection of Listeria monocytogenes. Chin. J. Lab. Diagn., 13: 1231-1234.
Direct Link - Moreno, Y., L. Ballesteros, J. Garcia-Hernandez, P. Santiago, A. Gonzalez and M.A. Ferrus, 2011. Specific detection of viable Listeria monocytogenes in Spanish wastewater treatment plants by fluorescent in situ hybridization and PCR. Water Res., 45: 4634-4640.
CrossRefDirect Link - Karoonuthaisiri, N., R. Charlermroj, J. Teerapornpuntakit, M. Kumpoosiri and O. Himananto et al., 2015. Bead array for Listeria monocytogenes detection using specific monoclonal antibodies. Food Control, 47: 462-471.
CrossRefDirect Link - Sergeev, N., M. Distler, S. Courtney, S.F. Al-Khaldi, D. Volokhov, V. Chizhikov and A. Rasooly, 2004. Multipathogen oligonucleotide microarray for environmental and biodefense applications. Biosens. Bioelectron., 20: 684-698.
CrossRefDirect Link - Wanatabe, S., S. Ito, Y. Kamata, N. Omoda and T. Yamazaki et al., 2001. Development of competitive Enzyme-Linked Immunosorbent Assays (ELISAs) based on monoclonal antibodies for chloronicotinoid insecticides imidacloprid and acetamiprid. Analytica Chimica Acta, 427: 211-219.
CrossRefDirect Link - Suo, B., Y. He, G. Paoli, A. Gehring, S.I. Tu and X. Shi, 2010. Development of an oligonucleotide-based microarray to detect multiple foodborne pathogens. Mol. Cell. Probes, 24: 77-86.
CrossRefDirect Link - Wang, H.Y., Z.X. Liu, X.H. Shi, L.L. Zhao, H. Liu and H.T. Zhen, 2006. [Research advancement of Listeria monocytogenes and its surface proteins]. Inspection Quarantine Sci., 2: 76-80, (In Chinese).
Direct Link - Wang, H., Z. Liu, H. Liu, L. Zhao and H. Zhen, 2006. Establishment of a rapid sensitive and specific PCR detection method of Listeria monocytogenes in food. Inspection Quarantine Sci., 1: 3-6.
Direct Link - Wang, Y.K., Y.C. Wang, H.A. Wang, W.H. Ji, J.H. Sun and Y.X. Yan, 2014. An immunomagnetic-bead-based enzyme-linked immunosorbent assay for sensitive quantification of fumonisin B1. Food Control, 40: 41-45.
CrossRefDirect Link - Xu, D.S., X.F. Wu and P.Q. Cheng, 2007. Comparison of real-time fluorescence PCR with PCR and bacterium culture in detection of Listeia monocytogenes. Chin. J. Health Lab. Tech., 17: 861-863.
Direct Link - Yan, B., Y.S. Li, G.C. Huo and Z.J. Jiang, 2006. Rapid detection of Listeria monocytogenes in food. Sci. Technol. Food Ind., 27: 202-205.
Direct Link - Zhu, R.F. and C.Y. Ye, 2007. Virulence factors of Listeria monocytogenes. Chin. J. Food Hygiene, 19: 158-162.
Direct Link