
Niken Satuti Nur Handayani
Laboratory of Genetics and Breeding, Faculty of Biology, Universitas Gadjah Mada, Jl. Teknika Selatan, Sekip Utara, Yogyakarta 55281, Indonesia Tel/Fax: +62-274-580839
LiveDNA: 62.16385
Nailil Husna
Laboratory of Genetics and Breeding, Faculty of Biology, Universitas Gadjah Mada, Jl. Teknika Selatan, Sekip Utara, Yogyakarta 55281, Indonesia Tel/Fax: +62-274-580839
Immanuel Sanka
Laboratory of Genetics and Breeding, Faculty of Biology, Universitas Gadjah Mada, Jl. Teknika Selatan, Sekip Utara, Yogyakarta 55281, Indonesia Tel/Fax: +62-274-580839
Pakistan Journal of Biological Sciences
Year: 2017 | Volume: 20 | Issue: 7 | Page No.: 343-349







ABSTRACT
Background and Objective: The α-thalassemia is an inherited blood disorder affecting quality and quantity of hemoglobin. It caused mostly by deletion of one or two α-globin genes and characterized by deficient production of α-globin chain in hemoglobin leading from mild anemia to lethal. The α-globin gene with partial deletion could reduce chain production or produce abnormal chain. Its effect depends on mechanism of chain production affected. This study aimed to analyze the effect of partial deletion in α-globin gene influencing the mechanisms to produce functional α-globin chain in α-thalassemia cases. Materials and Method: The three mutant genes from genebank were selected and processed. The analysis performed in deleted sequences determination, mRNA sequences, protein structures and protein chains interaction to form hemoglobin by SWISS MODEL, CHIMERA and SABLE Polyview 2D. Results: The result showed 76 amino acids deleted in one mutant α-globin gene (V00516.1). The mutation gave effect in every mechanism of the α-globin chain conformation and production. It affected protein conformation by losing over half the helical chains. It reduced the function completely, in which, disturb hemoglobin A (HbA) production with emergence of β-sheets conformation. Conclusion: The analysis concluded that the protein produced by the α-globin gene with partial deletion lost its function and unable to form hemoglobin.
PDF Abstract XML References Citation
Received: February 24, 2017;
Accepted: May 05, 2017;
Published: June 15, 2017
Copyright: © 2017. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Niken Satuti Nur Handayani, Nailil Husna and Immanuel Sanka, 2017. α-globin Alteration in α-thalassemia Disorder: Prediction and Interaction Defect. Pakistan Journal of Biological Sciences, 20: 343-349.
DOI: 10.3923/pjbs.2017.343.349
URL: https://scialert.net/abstract/?doi=pjbs.2017.343.349
DOI: 10.3923/pjbs.2017.343.349
URL: https://scialert.net/abstract/?doi=pjbs.2017.343.349
INTRODUCTION
Hemoglobin (Hb), a transport protein, has an important role in blood regulation. This protein consists of four heme groups on each globin chain. The oxygen from each heme group will bind ferrous ion1. There are several types of hemoglobin which can be formed from many different polypeptide chain components. Furthermore, eight types of hemoglobin already studied in embryonic, fetal and adult phase. In human body, a hemoglobin consists of 2 (two) pairs of globin chain (α-globin and β-globin) 2. There are six of eight types of Hb which are constructed by α-globin chain. One of them is hemoglobin A (HbA) that constructed by two α-globin chains with two β-globin chains3. The α-globin chain is expressed by two α-globin genes which located in chromosome number 164. However, these genes can be altered and formed a genetic disorder called α-thalassemia. The mutation causes an abnormality with low blood quality and quantity production5-6. The mutation of α-globin chain will disrupt the blood protein and ferrous ion regulation7. In the end, the abnormality changes the feature of normal hemoglobin within the whole function.
Almost all of the mutation types in α-thalassemia are formed by a deletion in the DNA sequence. The deletion affects the α-globin chain and results in the failure protein synthesis8. Unfortunately, thalassemia research is commonly stopped until the disorder detection through DNA sequence alteration. Nevertheless, the alteration of DNA sequence could give the more information about the mechanism of mutation defect. Nowadays, many researchers use the bioinformatics and bio-modelling to get the projection or prediction of the DNA alteration9.
This study aimed to analyze the effect of partial deletion in α-globin gene. It would provide the defect prediction from the expression of mutant gene. Massive alteration which possibly found providing the urgency of confirmation research in laboratory with molecular biology technique to test blood or serum obtained from α-thalassemia patients.
MATERIALS AND METHODS
Template collection: The GenBank (NCBI) database was used to determine the homology model. The nucleotides sequence which used to depict the α-globin mutant was V00516.1, V00492.1 and AF525460.1. Normal α-globin sequence NC_000016.10 was also acquired as a comparison. To get normal globin structure, protein with PDB ID 4hhb and PDB ID 1z8u were used10.
Sequence alignment: Multiple Sequence Alignment (MSA) was performed by using MUSCLE in MEGA6 to find the massive deletion11. From the MSA result, coding regions were determined then translated to amino acid sequences and submitted into protein structure prediction program.
Structure prediction and analysis: The prediction of protein was elucidated from SWISS-MODEL Expasy12. Superimposition was required for predicting the abnormality in CHIMERA. Moreover, SABLE Polyview 2D was accomplished to visualize the structure disruption of the mutant gene phenotype13.
RESULTS
The alignment of three mutants to the normal gene showed one mutant (V00492.1) with 171 nucleotides coding sequence deleted (Fig. 1a), mutation of the other two located in intervening sequences. Whole sequence of V00492.1 was aligned to HBA1 which showed two alternatives Open Reading Frame (ORF) besides deleted normal ORF (Fig. 1b). To test these two ORF, the amino acid sequences were aligned with α-globin protein. The alignment showed that using the first ORF and the translation would end immediately after four amino acid chain synthesized (Fig. 1c). Therefore for further analysis second ORF was used to obtain α-globin polypeptide.
The α-complex region of HBA2 located in 3’UTR precisely at nt25-nt70 with C rich regions (Fig. 2a). Mutant gene which generally analyzed is HBA1 with partial deletion in the upstream gene. The α-complex region of mutant genes predicted through alignment. The result showed same size and location but there is polymorphism on nucleotides. Generally, the HBA1 have less C nucleotides than HBA2 (Fig. 2b).
The modeling result showed mutant chain with only 66 amino acid and form three helices and four non-helical (Fig. 3a). Heme group attaches to proximal histidine (His12). The superimposition with normal protein showed deleted 75 amino acids which form four helical chains missing (Fig. 3b). Therefore amino acid order shifted ex: proximal histidine (His87 to His12).
The salt bridge interactions are between α-β chain and α-α chain. Mutant chains with short amino acid lost Val 1 and Lys 40 as charged amino acid which facilitates the salt bridge interaction with β chains. On mutant chains, only arginine and aspartic acid were found possibly to form salt bridge interaction (Fig. 4).
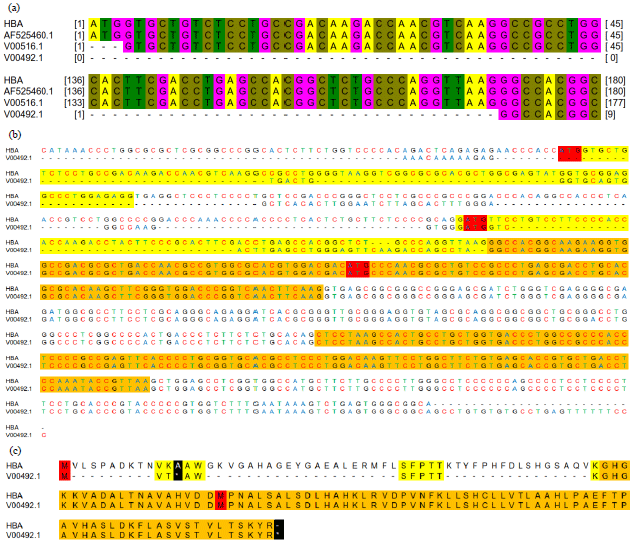 | |
| Fig. 1(a-c): | Sequences alignment of α-globin sequences, (a) Codon sequences alignment of three mutant genes, (b) Whole gene alignment of mutant gene V00492.1 with normal sequences. Two Open Reading Frames (ORF) colored red, coding sequence of normal gene colored yellow and orange, which is consist of three exons and coding sequence of mutant gene colored orange and only has two exons and (c) Amino acid alignment. Methionine colored red as ORF result. Predicted coding sequences trough upstream ORF colored yellow. Coding sequences from the GenBank colored orange. Translation trough upstream ORF stopped by stop codon (colored black) after 3 amino acid translated |
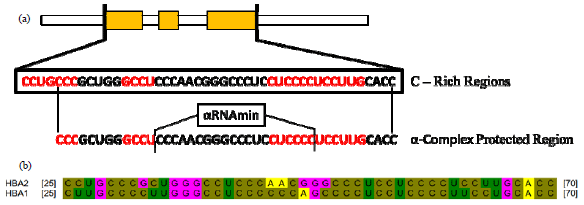 | |
| Fig. 2(a-b): | α-complex region, (a) α-complex of HBA2 modified from Waggoner and Liebhaber (2003) located in 3’UTR of HBA2 nucleotide 25-70 (after exon’s stop codon)14. The red boxes and red sequences represent the C-rich regions and (b) α-complex sequences alignment of HBA2 and HBA1 to determine α-complex in HBA1 and mutant gene. HBA1 also represent the mutant gene because both sequences showed same nucleotides |
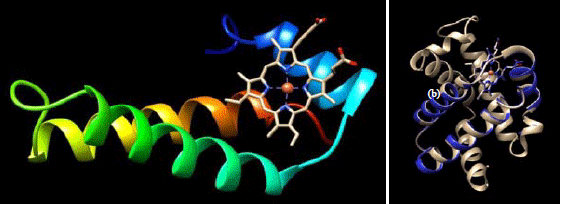 | |
| Fig. 3(a-b): | (a) α-globin mutant chain (template: PDB ID 1o1j) and (b) Superimposition mutant chain and normal chain of α-globin (template PDB ID 4hhb). Normal chain colored grey and mutant chain colored blue. The structure visualization used CHIMERA and superimposed with the MatchandMaker menu |
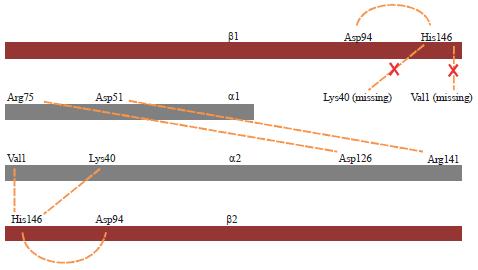 | |
| Fig. 4: | Salt bridge interactions modified with mutant prediction15, the orange line represents the salt bridge interactions to form hemoglobin. There are two interactions that could not be formed in mutant chain due to deletion |
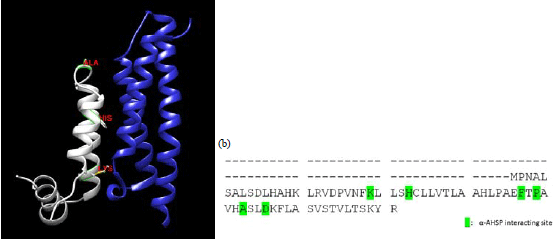 | |
| Fig. 5(a-b): | Alpha-Hemoglobin Stabilizing Protein (AHSP) binds to mutant chains, (a) Prediction through superimposition to the normal chain (template PDB ID 1z8u). AHSP molecules are shown in dark blue and mutant chain shown in white. The contact residue is shown with red label. Contact residues locate in G-helices and GH non-helical which still available in mutant chain and (b) α-AHSP interaction site of mutant chains16 |
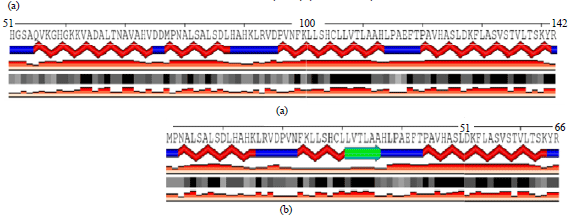 | |
| Fig. 6(a-b): | Two dimensions visualization (a) normal chain, amino acid 51-142 and (b) mutant chain, amino acid 1-66. α-helix is shown in colored red, non-helical chain colored blue and β-strand or bridge colored blue with arrow at the end. β-strand is only found in mutant chain. The box below shows relative accessible surface of each amino acid residue (white: completely buried, black: fully exposed) |
Modeling of mutant chain and AHSP performed to investigate the availability of contact residue in mutant gene. The mutant gene could bind with AHSP trough three basic contact residues which are Lys99, His103 and Tyr117 shifted to Lys24, His28 and Tyr42 in mutant gene (Fig. 5). Complete contact residue of mutant chain to AHSP showed in Fig. 5b with only one residue missed. It was Arg31 in the normal protein.
Secondary structure visualization between mutant and normal chain showed different structured appeared (Fig. 6). α-helix formed with 1-5 amino acid shifted in mutant chain compared to normal chain. β-strand appeared in mutant chain, specifically at amino acid 32-38. The differences also showed in the relative accessible surface indicated by different color in mostly amino acid.
DISCUSSION
Deletion in V00492.1 causes a premature translation termination through Open Reading Frame (ORF) in the upstream or first ORF. Translation reinitiation with downstream ORF generates a shorter polypeptide chain (66 amino acids) than the normal α-globin chain (142 amino acids). It is because there were 13 amino acids from the coding sequence that was not translated (Fig. 3c). This translation re-initiation might be caused by 5’ proximal nonsense mutation which may contribute to the alleviation of nonsense-mediated mRNA decay (NMD) and modulate disease phenotype16.
The coding sequence of α-globin mutant was 198 nucleotides translated with second ORF. This mutation could disrupt not only the translation but also the post-transcriptional mechanism. The mRNA stability is a critical determinant of normal globin development and its function. The α-complex is formed to stabilize the α-globin in the post-transcription and on the translation process14. The α-complex is located at the 3’UTR of the RNA with C-rich region and binds to PolyA binding protein, including prevents the mRNA decay mechanism. The α-complex of mutant gene depicted less C nucleotides. The α-complex structure was with C-Rich Regions that comprise the stability determinant. The sequences of the α-complex act as a Protected Region (PR) that contains the ErEN site and the minimal α-complex binding site (α-RNAmin)14. The α-complex region of HBA1 conformation was generated by alignment and mapping.
Mutant chain with heme group showed in 3d modeling was formed by an availability of the proximal histidine on the chain. Proximal histidine on charged F helixes in the normal chain initiates the heme-ionic binding interaction17. This proximal histidine was also shown on the mutant model. Furthermore, superimposition result showed the difference between normal model and mutant model. There are four helical chains which missing due to deletion. The deletion also caused absence of distal histidine in model. This absence erases the main function of distal histidine which is as an oxygen-transport protein in hemoglobin. The distal histidine also contributes to stabilize the protein while carrying oxygen on the protein18.
The deletion in mutant gene gave a huge effect in hemoglobin formation, particularly its structure from salt bridge interaction defect. Salt bridge interaction is used to stabilize the hemoglobin. The bridge between charged amino acid on four globin chains are used to make the ionic bond when the hemoglobin not capable of carrying oxygen. Or on the other word, it would be activated in deoxyhemoglobin state. Several amino acids are needed for initiating the salt bridge interaction19. By losing of the lysine and valine amino acids, the mutant chain would only form two of four interactions for the α-globin chain. Furthermore, α1 and β1 could not have salt bridge interaction to bind for one chain with another chain. Therefore the mutant gene was unable to form any hemoglobin.
Nonetheless, a hemoglobin conformation process also requires other protein interaction. One of the most important proteins which needed is a α-globin stabilizing protein (AHSP)20. The AHSP specifically bind the free α-globin before constructing a dimer with β-globin. The AHSP recognized the G and H helices of α-globin through the hydrophobic interface and form larger conform coupled to the α-β dimer. The AHSP and α-globin complex prevent the free α-globin precipitation and damage the blood cell membrane, this anomaly occurs in β-thalassemia3. Two dimers of the complex formed a hemoglobin molecule with ionic interaction within each α chain18. Nonetheless, a hemoglobin H (Hb-H) could be formed and caused by tetramer production of β-globin chains due to failure regulation of α-globin synthesis. This mutant is found in the individual with massive deletion of α-globin gene, which produces 3 out of 4 missing genes7.
In this case, the sequences of the gene exist and translated into amino acid trough re-initiation of the start codon in the downstream. The mutant chain could bind AHSP with the existed binding site. Regardless, it was assumed that the failure regulation occurs on imperfect dimer formation to produce hemoglobin. Dimer formation of α-β-globin used ionic interaction on charged amino acid which is missing in the mutant chain. Therefore it could not form dimer or tetramer hemoglobin. It assumed that the mutant chain would not disrupt membrane of erythrocyte because it was stabilized by AHSP. Next, investigation of complex mutant chain and AHSP in erythrocyte needed to know any hazard effect of its accumulation or continuous formation in the cell.
The secondary structure and its relative surface accessible interaction between a normal α-globin chain and mutant globin chain were identified by looking the secondary structure changes. The simulation explains the contact preferences of the local and non-local amino acid residue to stabilize the conformation21.One β-strand was found and formed on the mutant chain, which is not normally found on the normal α-globin chain. The β-sheet could be formed by hydrogen bonds between several β-strands 22. The β-strand rearranges α-helix structure and folding position by providing the hydrogen bonds. This interaction could affect the folding conformation of the α-globin mutant and resulted as abnormal chains. Relative Solvent Accessible Surface Area (RSA) contributed to determine protein folding process and predict the conformation changes upon the binding site23. The RSA shown in Figure indicate different folding which occurs on mutant and normal chain. It might also be caused by β-strand formation on mutant chain.
By conducting this study, the mutation alteration affected the α-globin chain, not only structurally but also functionally. From hereafter, this research provided the data to proceed for further analysis such as molecular dynamics and could give more information to interfere the effect of mutations in the α-globin. There are many deletions causing α-thalassemia that need to be analyzed its effect in hemoglobin production and its possibility to damage erythrocyte membrane which could increase severity of the disease.
CONCLUSION
In the α-globin mutant gene, 171 nucleotides of coding sequence had been deleted. The partial deletion affected each step of α-globin chain production from the mRNA synthesis to the protein binding to form hemoglobin. The remaining sequence could express and produce short chain with defective structure and completely lost its function and ability to form hemoglobin.
SIGNIFICANCE STATEMENT
The present study provides a picture of mutation that produces cascade reaction, not only the DNA alteration but also the protein folding and conforms. The α-globin mutant disrupts the function and structure of hemoglobin production. As such, this result could also strengthen the reason how the thalassemia disorder has the incapability to get the normal quality and quantity of hemoglobin or blood cell production.
ACKNOWLEDGMENTS
The authors would like to give a thank you remark to research grant from Operational Fund of States Universities of Faculty Biology Universitas Gadjah Mada, with grant number UGM/BI/1965/UM/01/34 that has given financial support to execute this study.
REFERENCES
- Marengo-Rowe, A.J., 2006. Structure-function relations of human hemoglobins. Proc. (Baylor Univ. Med. Center), 19: 239-245.
Direct Link - Vichinsky, E.P., 2009. Alpha thalassemia major-new mutations, intrauterine management and outcomes. Hematol. Am. Soc. Hematol. Educ. Progr., 2009: 35-41.
CrossRefDirect Link - Weatherall, D.J., 2008. Hemoglobinopathies worldwide: Present and future. Curr. Mol. Med., 8: 592-599.
CrossRefDirect Link - Ahmad, R., M. Saleem, N.S. Aloysious, P. Yelumalai, N. Mohamed and S. Hassan, 2013. Distribution of alpha thalassaemia gene variants in diverse ethnic populations in Malaysia: Data from the institute for medical research. Int. J. Mol. Sci., 14: 18599-18614.
CrossRefDirect Link - Harteveld, C.L. and D.R. Higgs, 2010. α-thalassaemia. Orphanet J. Rare Dis., Vol. 5.
CrossRefDirect Link - Forget, B.G. and H.F. Bunn, 2013. Classification of the disorders of hemoglobin. Cold Spring Harbor Perspectives Med., Vol. 3.
CrossRef - Feng, L., D.A. Gell, S. Zhou, L. Gu and Y. Kong et al., 2004. Molecular mechanism of AHSP-mediated stabilization of α-hemoglobin. Cell, 119: 629-640.
CrossRefDirect Link - Tamura, K., G. Stecher, D. Peterson, A. Filipski and S. Kumar, 2013. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol., 30: 2725-2729.
CrossRefPubMedDirect Link - Bordoli, L., F. Kiefeer, K. Arnold, P. Benkert, J. Battey and T. Schwede, 2009. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc., 4: 1-13.
CrossRefDirect Link - Porollo, A.A., R. Adamczak and J. Meller, 2004. POLYVIEW: A flexible visualization tool for structural and functional annotations of proteins. Bioinformatics, 20: 2460-2462.
CrossRefDirect Link - Waggoner, S.A. and S.A. Liebhaber, 2003. Regulation of alpha-globin mRNA stability. Exp. Biol. Med. (Maywood), 228: 387-395.
PubMedDirect Link - Yu, X., T.L. Mollan, A. Butler, A.J. Gow, J.S. Olson and M.J. Weiss, 2009. Analysis of human α globin gene mutations that impair binding to the α hemoglobin stabilizing protein. Blood, 113: 5961-5969.
CrossRefDirect Link - Thom, C.S., C.F. Dickson, D.A. Gell and M.J. Weiss, 2013. Hemoglobin variants: Biochemical properties and clinical correlates. Cold Spring Harbor Perspectives Med., Vol. 3.
CrossRef - Birukou, I., R.L. Schweers and J.S. Olson, 2010. Distal histidine stabilizes bound O2 and acts as a gate for ligand entry in both subunits of adult human hemoglobin. J. Biol. Chem., 285: 8840-8854.
CrossRefDirect Link - Peixeiro, I., A.L. Silva and L. Romao, 2011. Control of human β-globin mRNA stability and its impact on beta-thalassemia phenotype. Haematologica, 96: 905-913.
CrossRefDirect Link - Domingues-Hamdi, E., C. Vasseur, J.B. Fournier, M.C. Marden, H. Wajcman and V. Baudin-Creuza, 2014. Role of α-globin H helix in the building of tetrameric human hemoglobin: Interaction with α-hemoglobin stabilizing protein (AHSP) and heme molecule. PloS One, Vol. 9.
CrossRef - Guilloux, A., B. Caudron and J.L. Jestin, 2013. A method to predict edge strands in beta-sheets from protein sequences. Comput. Struct. Biotechnol. J., Vol. 7.
CrossRef - Marsh, J.A. and S.A. Teichmann, 2011. Relative solvent accessible surface area predicts protein conformational changes upon binding. Structure, 19: 859-867.
CrossRefDirect Link