
Fazlul Huq
School of Biomedical Sciences, Faculty of Health Sciences, The University of Sydney, Australia
Journal of Pharmacology and Toxicology
Year: 2007 | Volume: 2 | Issue: 3 | Page No.: 238-247







ABSTRACT
Irbesartan (IS) is a potent, long-acting receptor antagonist for the octapeptide angiotensin II (AII), having high selectivity for the AT1 subtype. AII accelerates the development of atherosclerosis by activating AII subtype 1 receptors that promote generation superoxide anion and cause oxidative stress, leading to activation of nuclear transcription factor and endothelial dysfunction. IS is used in the treatment of hypertension, diabetic nephropathy and heart failure. The drug is metabolized in animals and humans to give at least seven urinary metabolites (denoted as M1, M2, M3, M4, M5, M6 and M7) although it does not rely on biotransformation for its pharmacological effect. IS shows minimal potential for drug or food interactions. Molecular modelling analyses based on molecular mechanics, semi-empirical (PM3) and DFT (at B3LYP/6-31G* level) calculations show that IS and its metabolites have moderately large LUMO-HOMO energy differences ranging from 5.0 to 5.2 eV from DFT calculations, indicating that IS and all its metabolites will be kinetically inert. Thus, although the molecules have some electron-deficient regions on their surface so that they could potentially react with glutathione and nucleobases in DNA, the high kinetic inertness of the molecules is believed to provide protection against such adverse reactions.
PDF Abstract XML References Citation
How to cite this article
Fazlul Huq, 2007. Molecular Modelling Analysis of the Metabolism of Irbesartan. Journal of Pharmacology and Toxicology, 2: 238-247.
DOI: 10.3923/jpt.2007.238.247
URL: https://scialert.net/abstract/?doi=jpt.2007.238.247
DOI: 10.3923/jpt.2007.238.247
URL: https://scialert.net/abstract/?doi=jpt.2007.238.247
INTRODUCTION
Increase in obesity and type 2 diabetes and the resulting consequences in cardiovascular morbidity and mortality have become an issue of great concern among public health authorities worldwide (York et al., 2004; Zimmet et al., 2001; Pershadsingh, 2006). The metabolic syndrome (MetS) represents a cluster metabolic and cardiovascular risk factors that predispose the sufferers to heart attack, stroke, heart failure and sudden cardiac death (Pershadsingh, 2006). Chronic inflammation and an abnormal pro-oxidant state may play a significant role in the development of epithelial dysfunction and the progression of atherosclerosis (Ross, 1999) and the two may also play a role in the pathogenesis of MetS as both are found in the syndrome (Sola et al., 2005). Irbesartan (IS, SR47436; BMS-186295; 2-butyl-3-[[2’-(1H-tetrazole-5-yl)[1,1’-biphenyl]-4-yl]methyl]-1,3-diazaspiro[4,4]non-1-en-4-one) is a potent, long-acting receptor antagonist for the octapeptide angiotensin II (AII), having high selectivity for the AT1 subtype (Cazaubon et al., 1993). It is used in the treatment of hypertension, diabetic nephropathy and heart failure (Eberhardt et al., 1993; Gottlieb et al., 1993). Like telmisartan, it provides a novel approach in addressing the multifactorial components of MetS.
Angiotensin II accelerates the development of atherosclerosis by activating angiotensin II type 1 receptors that promote generation superoxide anion and cause oxidative stress, leading to activation of nuclear transcription factor and endothelial dysfunction (Koh et al., 2006).
IS is well absorbed both in the presence and absence of food and in co-administration of other drugs (Hong et al., 2005), with bioavailability of 60-80% (Unger, 1999). IS metabolized in animals and humans to give at least seven urinary metabolites (denoted as M1, M2, M3, M4, M5, M6 and M7) (Chando et al., 1998; Stearns et al., 1992; Davi et al., 2000), although it does not rely on biotransformation for its pharmacological effect.
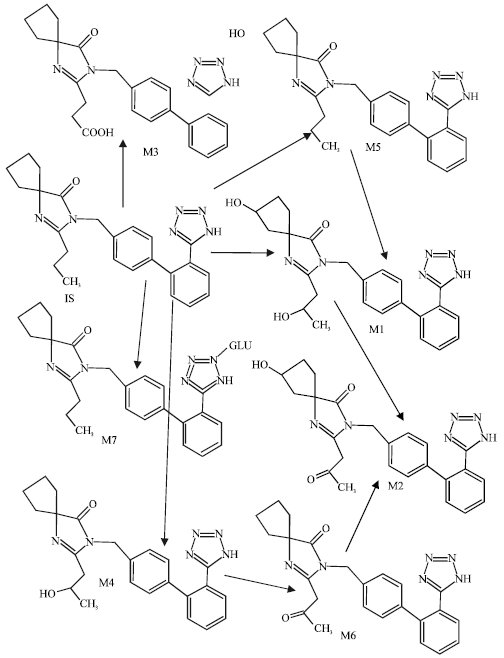 | |
| Fig. 1: | Metabolic pathways for irbesartan (Based on Chando et al., 1998) |
M1 is a tetrazole N2-β-glucuronide, M2 is obtained from monohydroxalyation of IS at the ω-1 (C3) position of the butyl side chain. M3 is the keto derivative of M2. M4 and M5 are two different monohydroxylated metabolites resulting from the oxidation of the spirocyclopentane ring. M6 and M7 are obtained oxidation of M4 and M5 respectively (Alexandre et al., 2004). CYP2C9 is the major cytochrome P450 isoform involved in the metabolism of IS as it is the case for other clinically important drugs such as phenytoin, warfarin and losarin (Cazaubon et al., 1993).
In this study, molecular modelling analyses have been carried out using the program Spartan ’02 (Spartan, 2002) to investigate the relative stability of IS and its metabolites with the aim of providing a better understanding on their relative toxicity. The work was carried out in the School of Biomedical Sciences, The University of Sydney during February to August 2006.
Computational Methods
The geometries of IS and its metabolites have been optimized based on molecular mechanics (Fig. 1), semi-empirical and DFT calculations, using the molecular modelling program Spartan ’02. Molecular mechanics calculations were carried out using MMFF force field. Semi-empirical calculations were carried out using the routine PM3. DFT calculations were carried at B3LYP/6-31G* level. In optimization calculations, a RMS gradient of 0.001 was set as the terminating condition. For the optimized structures, single point calculations were carried out to give heat of formation, enthalpy, entropy, free energy, dipole moment, solvation energy, energies for HOMO and LUMO. The order of calculations: molecular mechanics followed by semi-empirical followed by DFT ensured that the structure was not embedded in a local minimum. To further check whether the global minimum was reached, some calculations were carried out with improvable structures. It was found that when the stated order was followed, structure corresponding to the global minimum or close to that could ultimately be reached in all cases. Although RMS gradient of 0.001 may not be sufficiently low for vibrational analysis, it is believed to be sufficient for calculations associated with electronic energy levels.
RESULTS AND DISCUSSION
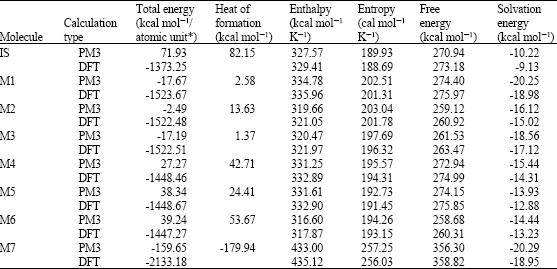
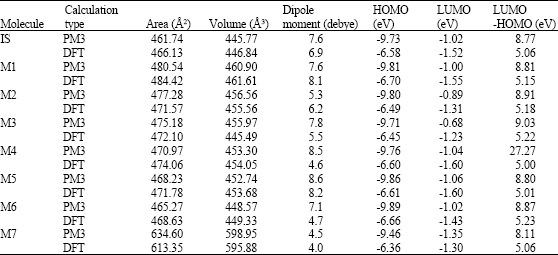
Table 1 gives the total energy, heat of formation as per PM3 calculation, enthalpy, entropy, free energy, surface area, volume, dipole moment, energies of HOMO and LUMO as per both PM3 and DFT calculations for IS and its metabolites M1, M2, M3, M4, M5, M6 and M7. Figure 2-9 give the regions of negative electrostatic potential (greyish-white envelopes) in (a) and density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) in (b) as applied to the optimized structures of IS and its metabolites M1, M2, M3, M4, M5, M6 and M7.
The calculated solvation energies of IS and its metabolites M1-M7 from PM3 calculations are -10.22, -20.25, -16.12, -18.56, -15.44, -13.93, -14.44 and -20.29, respectively and the corresponding dipole moments from DFT calculations are 6.9, 8.1, 6.2, 5.5, 4.6, 8.2, 4.7 and 4.0, respectively.
| Table 1: | Calculated thermodynamic and other parameters of IS and its metabolites |
 | |
| Table 1: | Continued |
 | |
| * in atomic units from DFT calculations | |
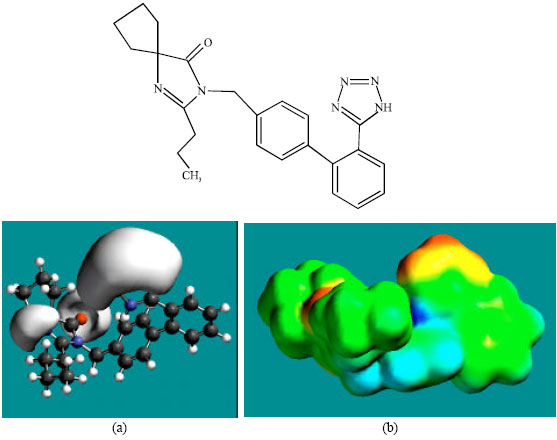 | |
| Fig. 2: | Structure of IS giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential) and in (b) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
The values suggest IS and its metabolites would not vary greatly in their solubility in water.
IS and its metabolites are found to have large LUMO-HOMO energy differences that range from 5.0 to 5.2 eV from DFT calculations, indicating that IS and all its metabolites would be kinetically inert.
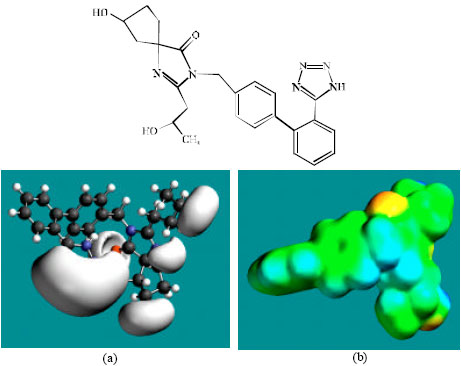 | |
| Fig. 3: | Structure of M1 giving in: (a) the electrostatic potential (greyish envelope enotes negative electrostatic potential) and in (b) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
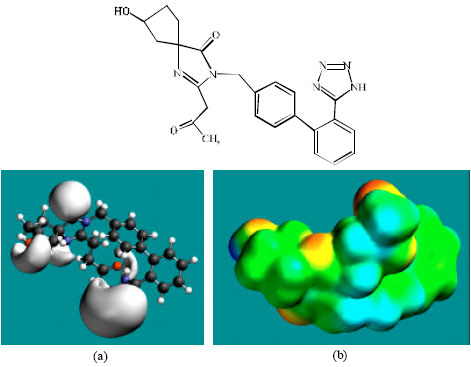 | |
| Fig. 4: | Structure of M2 giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential) and in (b) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
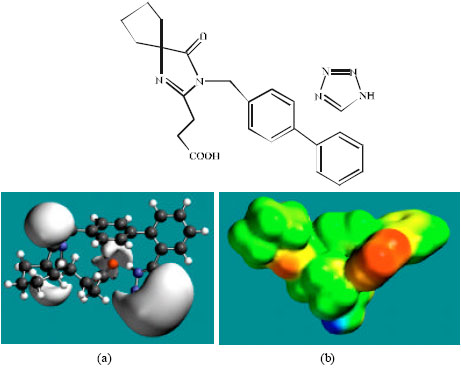 | |
| Fig. 5: | Structure of M3 giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential) and in (b) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
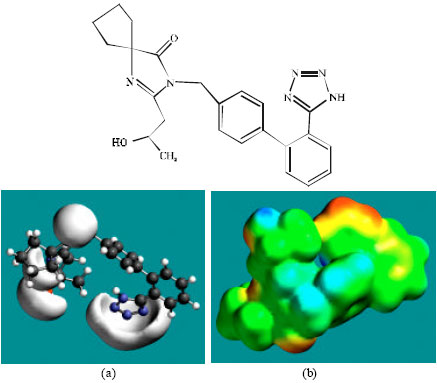 | |
| Fig. 6: | Structure of M4 giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential) and in (b) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
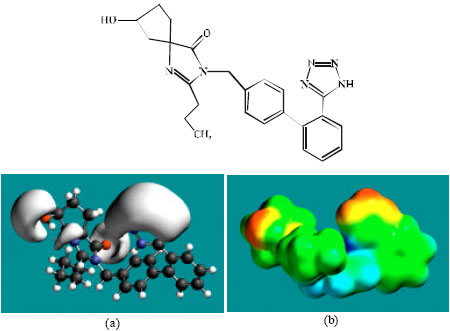 | |
| Fig. 7: | Structure of M5 giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential) and in (b) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
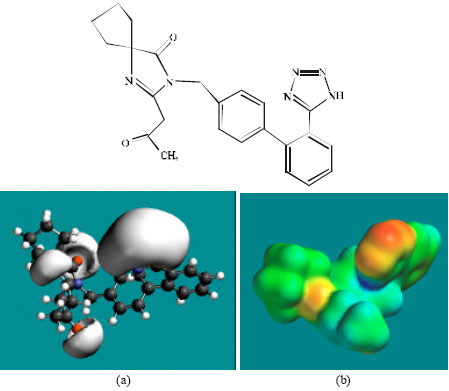 | |
| Fig. 8: | Structure of M6 giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential) and in (b) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
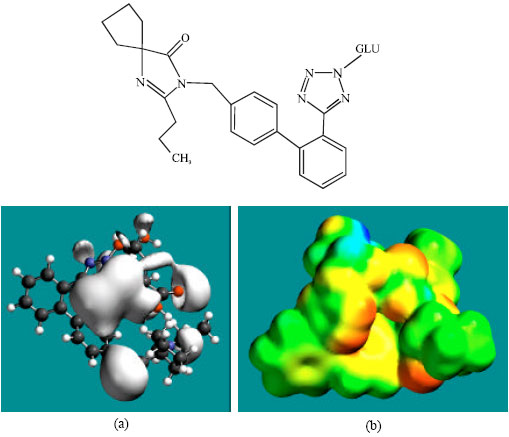 | |
| Fig. 9: | Structure of M7 giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential) and in (b) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
In the case of IS, M1, M2, M3, M4, M5, M6 and M7 the electrostatic potential is found to be more negative around the various nitrogen and oxygen centers, indicating that the positions may be subject to electrophilic attack.
Although not shown, when the positions of HOMos with high electron density and LUMOs are considered, the following points emerge. In the case of IS, the HOMOs with high electron density are found to be centered mostly on the non-hydrogen atoms of the 1,3-diazaspiro ring whereas the LUMO are centered on the non-hydrogen atoms of the tetrazole and phenyl rings. In the case of M1, M2, M3, M4, M5, M6 and M7, the HOMOs with high electron density are centered mostly on the non-hydrogen atoms of the 1,3-diazaspiro and the phenyl rings whereas the LUMO are centered mostly on the non-hydrogen atoms of the tetrazole and phenyl rings. The overlap of HOMO with high electron density and region of negative electrostatic potential close to sulfur, gives further support to the idea that the position may be subject to electrophilic attack.
The molecular surfaces of IS and its metabolites M1, M2, M3, M4, M5 and M6 are found to abound in neutral (green) regions indicating that the compounds generally would have low solubility in water. However, the compounds are also found to possess some negative (yellow and red) and positive (blue) regions indicating that they may be subject to electrophilic and nucleophilic attacks. Within the cell, the nucleophilic attack may be that due to glutathione and nucleobases in DNA. Reaction with glutathione will induce cellular toxicity by compromising the antioxidant status of the cell whereas that with nucleobases in DNA will cause DNA damage. However, as stated earlier, since IS and all its metabolites are expected to be kinetically inert, the rate of such adverse reactions may be low. The terminal metabolite M7 abounds almost equally in neutral green and negative yellow and red regions (and some positive blue regions) indicating that it would be most soluble in water.
When surface area and volume of IS and its metabolites M1, M2, M3, M4, M5, M6 and M7 are compared, it is found that the values for none of the metabolites match exactly with those of IS (with the closest match being found for M6) (Table 1), indicating that none of the metabolites may act as a substrate for the receptor to which IS binds. This may explanation why the pharmacological activity of IS is attributed to the parent drug and not to any of the metabolites.
CONCLUSIONS
Molecular modelling analyses based on semi-empirical and DFT calculations show that.IS and all its metabolites have large LUMO-HOMO energy differences so that they would be kinetically inert. This means that although all the compounds have some electron-deficient regions on the molecular surface so that they could potentially react with glutathione and nucleobases in DNA, in actual fact the rate of such adverse reactions may not be significant because of the kinetic inertness of the molecules.
ABBREVIATIONS
| IS: | Irbesartan; SR47436; BMS-186295; 2-butyl-3-[[2’-(1H-tetrazole-5-yl)[1,1’-biphenyl]-4-yl]methyl]-1,3-diazaspiro[4,4]non-1-en-4-one |
| MetS: | Metabolic syndrome |
| AII: | Angiotensin II |
| DFT: | Density functional theory |
| LUMO: | Lowest unoccupied molecular orbital |
| HOMO: | Highest occupied molecular orbital |
ACKNOWLEDGMENTS
Fazlul Huq is grateful to the School of Biomedical Sciences, The University of Sydney for the time release from teaching.
REFERENCES
- Alexandre, V., S. Ladril, M. Maurus and R. Azerad, 2004. Microbial models of animal drug metabolism Part 5. Microbial preparation of human hydroxylated metabolites of irbesartan. J. Mol. Catalysis B: Enzymat., 29: 173-179.
Direct Link - Cazaubon, C., J. Gougat, F. Bousquet, P. Guiraudou and R. Gayraud et al., 1993. Pharmacological characterization of SR 47436, a new nonpeptide AT1 subtype angiotensin II receptor antagonist. J. Pharmacol. Exp. Ther., 265: 826-834.
Direct Link - Chando, T.J., D.W. Everett, A.D. Kahle, A.M. Starret and N. Vachharajani et al., 1998. Biotransformation of irbesartan in man. Drug Metab. Dispos., 26: 408-417.
Direct Link - Eberhardt, R.T., R.M. Kevak, P.M. Kang and W.H. Frishman, 1993. Angiotensin II receptor blockade: An innovative approach to cardiovascular pharmacology. J. Clin. Pharmacol., 33: 1023-1038.
Direct Link - Gottlieb, S.S., K. Dickstein, E. Fleck, J. Kostis, T.B. Levine, T. LeJemtel and M. DeKock, 1993. Hemodynamic and neurochormonal effects of the angiotensin II antagonist losartan in patients with congestive heart failure. Circulation, 88: 1602-1609.
Direct Link - Hong, X., S. Zhang, G. Mao, S. Jiang and Y. Zhang et al., 2005. CYP2C9*3 allelic variant is associated with metabolism of irbesartan in Chinese population. Eur. J. Pharmacol., 61: 627-634.
Direct Link - Koh, K.K., M.J. Quon, S.H. Han, W.J. Chung, J.A. Kim and E.K. Shin, 2006. Vascular and metabolic effects of candesartan: Insights from therapeutic interventions. J. Hypertension, 24: S31-S38.
Direct Link - Pershadsingh, H.A., 2006. Treating the metabolic syndrome using angiotensin receptor anatagonists that selectively modulate peroxisome proliferator-activated receptor-γ. Int. J. Biochem. Cell Biol., 38: 766-781.
Direct Link - Ross, R., 1999. Atherosclerosis is an inflammatory disease. Am. Heart J., 138: S419-S420.
Direct Link - Sola, S., M.Q.S. Mir, F.A. Cheema, N. Khan-Merchant and R.G. Menon et al., 2005. Irbesartan and lipoic acid improve endothelial function and reduce markers of inflammation in the metabolic syndrome: Results of the irbesartan and lipoic acid in endothelial dysfunction (ISLAND) study. Circulation, 111: 343-348.
CrossRefDirect Link - Stearns, R.A., R.R. Miller, G.A. Doss, P.K. Chakravarty, A. Rosegay, G.I. Gatto and S.H.L. Chiu, 1992. The metabolism of DuP 753, a nonpeptide angiotensin II receptor antagonist, by rat, monkey and human liver slices. Drug Metab. Dispos., 20: 281-287.
Direct Link - Unger, T., 1999. Significance of angiotensin type I recptor blockade: why are we angiotensin II receptor blockers different? Am. J. Cardiol., 84: 9S-15S.
Direct Link - York, D.A., S. Rossner, I. Caterson, C.M. Chen and W.P. James et al., 2004. Americn Heart Association. Prevention Conference VII: Obesity, a world-wide epidemic related to heart disease and stroke. Group I. Worldwide demographics of obesity. Circulation, 110: e463-e470.
Direct Link - Zimmet, P., K.G.M.M. Alberti and J. Shaw, 2001. Global and societal implications of the diabetes epidemic. Nature, 414: 782-787.
CrossRefPubMedDirect Link