
R. G. Ahmed
Department of Zoology, Faculty of Science, Cairo University,
Beni-Suef Branch, Beni-Suef, Egypt
International Journal of Zoological Research
Year: 2006 | Volume: 2 | Issue: 2 | Page No.: 150-168







ABSTRACT
The number of reports on the effects of various types of radiation is gradually increasing because of weakening of the immune system. Radiation can penetrate into living cells and result in the transfer of radiation energy to the biological material. The absorbed energy can increase the reactive oxygen species and break chemical bonds and cause ionization of different biologically essential macromolecules, such as DNA membrane lipids and proteins. Damage to the cellular membrane release the hydrolytic enzymes responsible for various catabolic processes in the tissues and leads to cell death. An understanding of the pattern in critical cellular structures such as DNA is an important prerequisite for a mechanistic assessment of primary radiation injury. The DNA damage induced by radiation such as base alterations, cross linking, strands breaker chromosomal aberration which may in turn lead to mutations. In order to further explore the harmful effects of radiation. I have produced a variety of effects of radiation on the apoptosis and necrosis. Indeed, the present review has shown that the increase in the oxidative stress (increased endogenous production of the free radicals) due to radiation may be a reason for such a damage of the cell membrane, and may lead to harming the cellular elements (such as DNA). Here, one can hypothesize that, the cells with increased sensitivity to oxidative stress may be more susceptible to damage by radiation compared to normal cells. The ultimate biological consequences of this effect are subsequently processed by these cells. Much work remains to be done to firmly establish this concept.
PDF Abstract XML References
How to cite this article
R. G. Ahmed, 2006. Damage Pattern as a Function of Various Types of Radiation. International Journal of Zoological Research, 2: 150-168.
DOI: 10.3923/ijzr.2006.150.168
URL: https://scialert.net/abstract/?doi=ijzr.2006.150.168
DOI: 10.3923/ijzr.2006.150.168
URL: https://scialert.net/abstract/?doi=ijzr.2006.150.168
INTRODUCTION
The damage to a living cell by radiation takes place on a molecular level (Glasstone and Jordan, 1981). It may occur either directly by the release of energy, within the structure of the molecule itself or indirectly by the formation of highly reactive free radicals which interact with subcellular constituents. Membrane can be easily damaged by the peroxidative decomposition of their phospholipids. In addition, a mechanism exists whereby such damage of one membrane system can lead to the subsequent injury of other membrane physically separated from the intial locus (Farber, 1982).
Irradiation produces physical and chemical damage to tissues leading to cell death. Ionizing radiation interacts with cells transferring energy to molecular systems in discrete quanta and causes tissue damage through different simultaneous pathways, including reactive oxygen species (ROS) induced oxidative damage (Guelman et al., 1996; 2001). Also, development of common endpoints such as apoptosis due to radiation, which may diverse the initial injurious events, e.g., by direct destruction of DNA (Ojeda et al., 1994; Blank et al., 1997; Caricchio et al., 1998; Khodarev et al., 1998; Meng et al., 1998. Radiation also may induced genetical instability (Kadhim et al., 1995). On the other hand, opposite to ionizing radiation, the energy of microwave and radiofrequency do not transfer to biological macromolecules by the formation of chemically reactive ion pairs and free radicals (Somosy, 2000).
Thus, the following is a brief concerted attempt to describe the backgrounds and recent findings that provide specific suggestions for explore the deleterious effect of various types of radiation exposure.
DNA Damage Caused By Radiation
The biological effects from genotoxic agents such as ionizing radiation, UV radiation or alkylating agents are often postulated to be linearly linked to dose, i.e., not showing a threshold even at lowest exposure levels (Burkart et al., 1999). Such hypotheses are based on the notion that critical lesions in the genome might already be caused by a single radiation which induce ionization in or near DNA, or by covalent addition of reactive molecules to DNA. This notion has some merits, not least its simplicity and a linear term in dose-effect relationships might indeed be important for most endpoints involving single step mechanisms. In addition, the localized complex damage is much more efficient in creating fixed DNA damage leading to cellular malfunction than simple lesions (Fox and Prise, 1993; Goodhead, 1994). Moreover, Tsoulou et al. (2005) deduced that the radiation damage to DNA is not a spontaneous effect but rather is an ensemble of damaging events that occur asynchronously.
The biological target of radiation within the cell is DNA. Damage of radiation to DNA is caused through two pathways, direct effect and indirect effects are seen in Table 1.
| Table 1: | The pathways of radiation on damaging DNA |
 | |
Effect of UV on DNA
UV radiation induces a much wider range of DNA damage, such as protein-DNA crosslinks, single strand breaks and thymine glycol. A number of studies using UVC radiation (254 nm) have been conducted to demonstrate the effect of UV on DNA (Mitchell et al., 1991), since UVC is close to the maximum absorption spectrum of DNA to initiate photochemical reactions and is also available as nearly monochromatic light source. Moreover, DNA is not a chromophore for UV-A radiation (Rosenstein and Mitchell, 1987), but could be damaged by photosensitization reaction initiated through absorption of UV-A by unidentified chromophore. Purine photoadducts have recently attracted the attention of photobiologists because oxidation of guanine occurs upon exposure of isolated DNA to UVC and UVB light, in the absence of a photosensitizer (Ichihashi et al., 2003). This may be accounted for by the formation of singlet oxygen from the triplet state of purine and pyrimidine bases, only a contribution of an electron transfer at the nucleoside level (Ito and Kawanishi, 1997). Electron transfer or singlet molecular oxygen produced by UVB and UVA radiation targets DNA base guanine, giving rise to 8-hydroxydeoxyguanosine (8-OHdG) in the strand DNA (Cadet et al., 2000). 8-OHdG (Fig. 1) is shown to be a ubiquitous maker of oxidative stress (Kasai and Nishimura, 1984) since it can be generated under various agents including peroxynitrite .OH radical, one-electron oxidation oxidants and singlet oxygen. 8-OHdG is a miscoding lesion causing G to T transversion although it represents only a minor fraction of UV-induced mutation, even in the UVA region (Cheng et al., 1992). UV radiation also induces a much wider range of DNA damage, such as protein-DNA crosslinks, single strand breaks and thymine glycol.
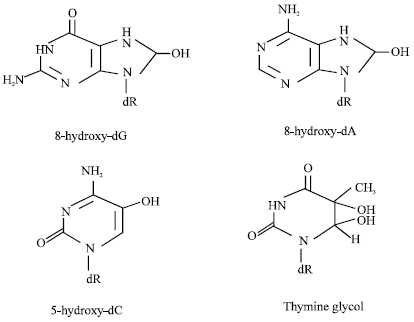 | |
| Fig. 1: | The major DNA lesions produced by oxidative damage, 8-oxoguanine, thymine glycol, pyrimidine hydrates and urea |
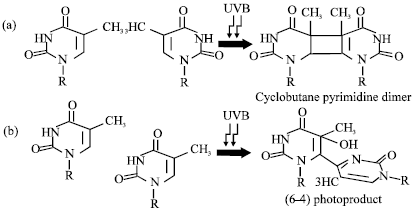 | |
| Fig. 2: | Adjacent pyrimidine bases on the same stand pyrimidine dimer (Fig. 2a) or (6-4) pyrimidine-pyrimidone photo product (Fig. 2b) after absorbing UVB light energy |
On the other hand, the effects of UVB radiation on DNA are mostly caused by the formation of dimeric photoproducts between adjacent pyrimidine bases on the same strand (Fig. 2, Ichihashi et al., 2003). The major class of lesions produced is cis-syn CPDs (cyclobutane pyrimidine dimers). Pyrimidine-pyrimidone (6-4) photoproducts ((6-4) pp) are the second most prevalent adducts formed in DNA by UVB radiation (Mitchell, 1988; Clingen et al., 1995). The yield of (6-4) pp is 5-10-fold less than that of CPDs (Eveno et al., 1995). (6-4) pp are converted to Dewar isomers by UVB rediation. (6-4) Adducts exhibiting a cytosine at their 5'-end may undergo deamination. The number of CPDs formed in a basal cell of human epidermis after exposure to three minimal doses of solar light about 60 min exposure at noon in the summer in Kobe, Japan (34° N) is calculated to be approximately 100000 per cell (Ichihashi et al., 1998). Among CPDs, thymine-cytosine (TC) and cytosine-cytosine (CC) dimers are shown to be the most mutagenic, since TC -»TT and CC -»TT mutations are frequently found in the p53 gene of UV-induced cancer cells (Daya-Grosjean et al., 1995). In the case of TT dimers, the major UV-induced photoproducts in humans are poorly mutagenic, since DNA polymerase preferentially incorporates 'A' residue opposite to non-instructional lesions, restoring the original seguence (A-T). (6-4) pp may be less mutagenic, since it is repaired efficiently compared to CPDs. Actually, 90% (6-4) pp are shown to be repaired at 3 h after irradiation (Nakagawa et al., 1998). In addition to pyrimidine photoproducts and adenine residue, which can either dimerize with an adjacent adenine or add to an adjacent thymine upon exposure to UVB radiation. The quantum yields of these photoproducts are very low, but these may contribute to the biological effects of UV light, since adenine-thymine adduct is shown to be mutagenic (Zhao and Taylor, 1996). Moreover, the ionization effect of ultraviolet radiation, especially, the ultraviolet B (UV-B), can be absorbed by nucleic acids and by some aromatic amino acids and may cause breaks and/or perturbation of molecular structures (Shimmura and Tsubota, 1997; Caricchio et al., 1998).
Furthermore, Tsoulou et al. (2005) recorded that the neutrons (high-linear energy transfer, H-LET) were more effective than gamma rays (low-linear energy transfer, L-LET) in inducing DNA, as well as a single electrons, were able to create clustered damage with a biologically relevant yield (Goodhead, 1994). However, high-LET charged particle radiation has several potential advantages over X-rays (Blakely and Kronenberg, 1998) such as, an excellent dose distribution, a higher relative biologic effectiveness (RBE), a reduction in the oxygen enhancement ratio, less variation in cell cycle-related radiosensitivity and less capability for repair of radiation injury.
UVA Damage To DNA In Cell-Culture Models (Wang et al., 2001)
UVA radiation has been shown to cause mutations in mammalian cells. Studies in murine cell lines have demonstrated that broadband UVA can induce mutations (Lundgren and Wulf, 1988). In addition, monochromatic UVA at 365 and 334 nm are mutagenic to a human epithelial P3 cell line (Jones et al., 1987) and to a Chinese hamster ovary cell line (Wells and Han, 1984) respectively. Enninga et al. (1986) have demonstrated that radiation at 365 nm was potentially mutagenic to cultured human fibroblasts. Wenczel et al. (1998) measured DNA single-strand breaks in cultured human melanocytes after UVA exposure. They showed that pheomelanin or melanin intermediates (or both) were the most likely chromophores that react with the UVA radiation, leading to the DNA single-strand breaks.
Using monochromatic radiation, Kvam and Tyrrell (1997) demonstrated that the wavelengths causing almost all of the oxidative DNA base damage (e.g., 7,8-dihydro-8-oxoguanine) in a human skin fibroblast cell line were in the UVA and visible light range. In addition, they estimated that the total amount of such guanine base damage induced by sunlight in fibroblasts of the skin equals or exceeds the amount of the major type of direct DNA damage, cyclobutane pyrimidine dimers induced mainly by UVB. Drobetsky et al. (1995) have characterized a specific mutation at the adenine phosphoribosyltransferase locus in Chinese hamster ovary cells irradiated with UVA. They demonstrated a high frequency (≤50%) of T to G transversion, a rare class of mutations ("fingerprint" mutation), in UVA-irradiated cells, compared with 9 in UVB-irradiated cells. A moderately high frequency (25%) of T to G transversion was seen with cells irradiated with simulated sunlight, leading to their conclusion that most of the T to G transversion mutations in cells irradiated with simulated sunlight can be attributed to the UVA portion of sunlight, with little, if any, contribution from UVB.
In addition to the aforementioned studies with different mammalian cell lines, the potential carcinogenic effect of UVA has also been demonstrated in cultured human melanocytes. Marrot et al. (1999) induced DNA breaks in the nucleus of Caucasian human melanocytes with broad-spectrum UVA (320-400 nm) irradiation. DNA breakage in cells was assessed via the comet assay, a technique used extensively for analyzing genotoxic effects by environmental chemicals (Belpaeme et al., 1998) or by UV components of the solar spectrum (Arlett et al., 1993; Alapetite et al., 1996). The investigators suggested that the endogenous pigments and/or melanin-related molecules seem to enhance DNA breakage after UVA irradiation. This is evidenced by higher DNA breakage in melanocytes than in fibroblasts, in cells with higher melanin content and in cells stimulated for melanogenesis by culturing in tyrosine-rich medium. In addition, Marrot et al. (1999) demonstrated that there was an increased level of p53 expression in the irradiated melanocytes, indicating DNA damage. That different UVA wavelengths induce mutations in different cell lines can be explained by the different types and amounts of photosensitizers or chromophores in these cells. Unlike UVB, UVA must first react with endogenous photosensitizers (eg, flavins, porphorins, melanins) that in turn generate reactive oxygen species that cause single-strand breaks or photoadducts. The different sensitivities and mechanisms for DNA damage induced by UVA and by UVB are important to elucidate to improve the formulation of sunscreens with broad-spectrum coverage.
Because the most effective wavelength in causing photoproduct formation in human skin is around 300 nm in the UVB spectrum (Freeman et al., 1989), efforts could be focused on developing UVB-protective sunscreens with maximal protection at or around this wavelength. However, the various photosensitizers in skin have different absorption spectra. These photosensitizers absorb different spectral bands of UVA and then generate oxygen radicals. Thus for UVA-protective sunscreens to be maximally effective, they should have broad coverage across the entire UVA spectrum.
Chromosomal Aberrations Due to Radiation
Gaps and breaks within the chromatid strands and chromosomes are the most frequently observed radiation-induced chromosomal lesions (Evans, 1962; Yu, 1971; Brecher, 1977). Combined light and electron microscopic studies by several authors (Humphrey and Brinkley, 1969; Brinkley and Hittelman, 1975; Brecher, 1977) revealed that radiation-induced gap-formation were a result of the physical loss of DNA-protein complexes rather than decondensation of the chromatin. Chromosome abnormalities, such as translocations, fragile tips and deletions were detected by high resolution scanning electron microscopy in canine chromosomes after whole-body irradiation (Niiro and Seed, 1988). Several studies on homologous chromosomes in the prophase of the first meiotic division showed the presence of breaks and rearrangements of synaptonemal complexes in irradiated cells (Backer et al., 1991; Alien et al., 1994; Johannisson et al., 1994). The ultraviolet radiation, especially UV-B absorbed by nucleic acids may also cause breaks or perturbation of the molecular structures of nucleic acids, manifested as chromosome aberrations (Shimmura and Tsubota, 1997; Caricchio et al., 1998).
A marked increase in the frequency of chromosome aberrations and formation of micronuclei was also observed in human blood lymphocytes upon 2450 MHz microwave exposure (Maes et al., 1993). This cytogenetical effect seems to be dependent on the frequency used, because it was absent in cells exposed to 935.2 MHz microwave irradiation (Maes et al., 1997).
These lesions can be caused directly by radiation energy and they can develop as a consequence of radiation induced genomic instability (Kadhim et al., 1995; Morgan et al., 1996) in the surviving cells. Measurement of chromosome aberrations is an informative and widely used technique for studying the genetic risk of ionizing radiation and is a useful biological indicator of radiation exposure.
On the other hand, low doses of irradiation are frequently followed by the formation of the so-called nuclear bodies, which appear ring-like chromatin aggregates surrounded by a less dense halo (Klein-Szanto et al., 1974; Barham and Walters, 1978; Orkisz and Bartel, 1981; Skog et al., 1983; Somosy et al., 1985). Their development may be accompanied by an increase in the processing or transport of pre-mRNA and/or pre-rRNA (Roth, 1995). The number of nucleoli and the average volume of nucleolar organizer regions may increase significantly upon irradiation, as reported by Morales et al. (1996). These phenomena may be related to the radiation-induced elevation of transcriptional activity of the nucleus. Irradiation may cause the formation of patches of dense substances in the nucleoli (Mironescu and Dragomir, 1967; Orkisz and Bartel, 1981). Nuclear segregation was also observed in irradiated cells (Klein-Szanto et al., 1974; Skog et al., 1983; Somosy et al., 1985).
Radiation Induced Reactive Oxygen Species
An "oxidative stress" should be considered as the consequence of a imbalance between pro-oxidant processes and effective antioxidant defense systems and finally the result of a detrimental disturbance of the cellular redox homeostasis (Meloni and Nicolay, 2003). Active oxygen species are generated during pathophysiologic conditions such as inflammation and ionizing radiation exposure (Kuo et al., 1993). Radiation-induced free radicals in turn impair the antioxidative defence mechanism, leading to an increased membrane lipid peroxidation (LPO) which results in damage of the membrane-bound enzymes (Halliwell and Gutteridge, 1989). Ultraviolet radiation generates reactive oxygen intermediates (ROIs) (Takahashi et al., 2000). Moreovere, in the human skin, the ultraviolet radiation causes increase of production of the reactive oxygen species which are responsible for oxidative stress and damage of many important cellular components in the target tissue (Firkle et al., 2000). Radiation-induced peroxidative damage determined in terms of the formation of thiobarbituric acid-reactive substances (TBARS) and the change in the specific activity of lactate dehydrogenase support this possibility (Srivastava and Kale, 1999). Lipid peroxidation increased as a function of radiation dose, from 45 to 600 Gy and the radiation-induced depletion of protein thiols (Kamat et al., 2000). Since lipid peroxidation and ionizing radiation are well known sources of free radicals and a damage of tissues.
Free radicals are highly reactive molecules generated by biochemical redox reactions that occur as part of normal cell metabolism and by exposure to environmental factors such as UV light and γ-radiation. In addition, acute ultraviolet-B (UV-B) irradiation is known to act as an initiator in the formation of reactive oxygen species. These oxygen products are highly reactive and they are able to cause irreversible damage to cellular components. The iron, singlet oxygen and hydrogen peroxide are important redox active species involved in the deleterious effects of UVA radiation on lipids and proteins of human skin cells (Vile and Tyrrell, 1995). Thus, cells with increased sensitivity to oxidative stress are more susceptible to damage by ionizing radiation than normal cells (Katz et al., 1996). In addition, the reactive oxygen species are believed to be involved in radiation lethality (Mansur et al., 2001). The involvement of oxidative stress in the UV irradiation-induced caspase activation and the subsequent apoptotic biochemical changes (Chan and Yu, 2000).
On the other hand, the radiofrequency fields of cellular phones may affect biological systems by increasing free radicals, which appear mainly to enhance lipid peroxidation, thus leading to oxidative stress (Moustafa et al., 2001). These results support the hypothesis that increased oxy-radical activity is a persistent effect in X-irradiated mammalian cells (Rugo and Schiestl, 2004).
Several mechanisms of ROS production have been described, including the mitochondrial respiratory chain, the multienzymatic cytochrome P450 complex in detoxication pathways, the xanthine oxidase, the nitric oxide (.NO) synthesis from arginine by .NO synthase and many cellular oxidases, such as cyclo-oxygenase or prolyl-hydroxylase for example (Nguyen et al., 2005). ROS are also generated from H2O2 or ozone in the presence of metal ions (iron or copper) through the Fenton's reaction or by various toxic compounds (Stohs and Bagchi, 1995). During inflammation, which may be induced by irradiation, superoxide anions are produced by various cell types, including white blood cells [polymorpho-nuclear neutrophils (PMN), macrophages or monocytes], endothelial cells or some parenchymal cells, through the activation of NADPH-oxidase, a cell membrane-associated enzymatic complex (Vignais, 2002).
Characterization of Radiation-induced Cell Death
Cell death includes apoptosis and necrosis. Cell death can be the ultimate consequence of cellular radiation injury (Okada, 1970a, Harms-Ringdahl et al., 1996; Blank et al., 1997; Hendry and West, 1997). Cell death caused by ionizing irradiation has previously been categorized into two main classes, based upon the time of disintegration of cells after exposure, namely into as interphase death and reproductive or mitotic death (Okada, 1970a). Interphase death is denned as an irreversible impairment of cellular metabolism and breakdown of cellular structures before entering into the first mitosis after irradiation. Reproductive or mitotic death occurs during mitosis and one or even several divisions after the irradiation (Okada, 1970b). Both interphase and reproductive death can be manifested as apoptosis and/or necrosis (Akagi et al., 1993; Nakano and Shinohara, 1994; Szumiel, 1994; Harms-Ringdahl et al., 1996).
The category of cell death depends on the type of cells and tissues and on the conditions and types of ionizing irradiation (Harms-Ringdahl et al., 1996; Blank et al., 1997; Hendry and West, 1997; Olive and Durand, 1997). Furthermore, the dose of irradiation may also play a role in determining the type of cell death. Moreover, recent researches have shown that the exposure to radiation results in induction of apoptosis in various mammalian cells (Wyllie et al., 1980; Yamada and Ohyama, 1988; Warters, 1992; Fuks et al., 1994; Haimovitz-Friedman et al., 1994; Szumiel, 1994; Stapper et al., 1995; Olive and Durand, 1997; Okunieff et al., 1998; Ramakrishnan et al., 1998; Radford, 1999; Lesnikov et al., 2001; Schwarz et al., 2002) and many observations suggest that apoptosis is the main form of ionizing radiation-induced cell death in lymphocytes, thymocytes, lymphoid and myeloid cell lines. However, some cell types show a slow and delayed apoptotic response (Hendry and West, 1997; Olive and Durand, 1997). The process starts within minutes following irradiation and lasts for several hours. UV-B irradiation may also induce apoptosis in several cell systems (Caricchio et al., 1998; Gniadecki et al., 1998). Apoptosis appears to be a critical cellular event in neuro-degenerative diseases (Adams et al., 1996; Michel et al., 1999) and in response to a variety of insults including cerebral ischemia and exposure to ionizing radiation (Linnik et al., 1995; Gobbel et al., 1998).
Apoptosis and classical necrosis are two genetically, biochemically and morphologically different types of cell death and the differences have been recognized as depending not only on the cell type but also on the radiation dose. The Table 2 shows the difference between apoptosis and necrosis under effect of radiation according the morphological criteria.
| Table 2: | The variation consequence between apoptosis and necrosis due to radiation exposure |
 | |
 | |
Indeed, Programmed cell death plays an important role in embryogenesis and can also be induced in different situations such as growth factor deprivation, chemical insults, hyperthermia and exposition to ultraviolet (UV) and ionizing radiation (Guo et al., 1997). More studies are necessary to further characterize the pathways involved in the gamma radiation induced apoptosis in embryos.
Radiation Induced Lysosomes and Their Processes
Pertaining to Table 3 Somosy (2000), the ionizing radiation caused the number and volume fraction of lysosomes to increase in the cells and elevated lysosomal enzyme activity can be detected by several authors (Brandes et al., 1967; Hamberg et al., 1977; Hamberg, 1983; Piao et al., 1983; Somosy, 2000; Rahman, 1963; Sottacasa et al., 1965; Harris, 1967; Zyss and Kaszczynska, 1972; Shah and Gadhia, 1979). At the same time, lysosomal enzymes may appear in the cytosol and in the extracellular fluid (Brandes et al., 1967; Harris, 1970; Rene et al., 1971; Conti et al., 1974; Snyder and Ekiund, 1978).
Moreover, some of the observations suggesting increased lysosomal enzyme activity in the irradiated cells are based on the fact that the volume fraction of lysosomes/autophagic vacuoles is increased in these cells. As well as, the expansion of the lysosomal/autophagic compartment may be interpreted with several caution; 1) may be due to a decreased rate of digestion of the segregated material, which may cause the overload and expansion of the lysosomal compartment (Somosy, 2000), 2) may be due to the accumulation of undegraded material as a consequence of the decreased rate of lysosomal protein degradation in the irradiated cells (Somosy et al., 1996), 3) cells such as macrophages and glial cells may respond to irradiation by elevated pinocytotic and phagocytic activity (Barham and Walters, 1978; Somosy et al., 1985; Gasperin et al., 1992), 4) lysosomes are extremely sensitive to radiation, peroxides and free radicals and that the damage resulting to the membrane can readily release the hydrolytic enzymes responsible for various catabolic processes in the tissues (Desal et al., 1964) and 5) the radiation induces physical or functional changes in the lysosomal membranes permitting release of the hydrolytic enzymes and indirectly causing death by this mechanism (Bacq and Alexander, 1961).
| Table 3: | Ionizing radiation-induced alteration of autophagic vacuole/lysosomal compartments in various cells (compiled data according to Somosy (2000) |
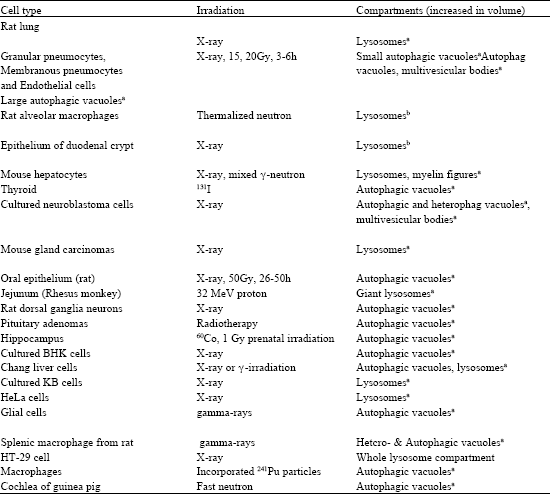 | |
| aQuantitative evaluation, bQualitative evaluation | |
There is an ample evidence that the lysosomal enzymes play an important role in the cellular physiologic and pathologic regressive changes including necrobiosis and cell death (Tanaka, 1979). Ionizing radiation appears to provoke the release and activation of lysosomal enzymes (Brandes et al., 1967). Following X-irradiation injury elevated levels of lysosomal enzymes, has been reported for a variety of tissues (De Duve, 1964; Goutier and Bacq, 1963). Also, the higher acid phosphatase activity noticed in the ground cytoplasm in many cases of cell injury is (Khattab, 1967; Khattab et al., 1970) due to the rupture of lysosomal membranes followed by the release of their contents of powerful enzymes into the cytoplasm where they induce their lytic effects. A high acid phosphatase activity is often associated with cell degeneration (Novikoff, 1961; Khattab, 1967) and the lysosomal rupture preceeds rather than results from cell death (De Duve, 1961). According to the above results, it is worth mentioning that, the framework of the "enzyme release" hypothesis of post-radiational cell death (Alexander and Bacq, 1961), which suggests that cell death is a consequence of the unregulated release of digestive enzymes from lysosomes after irradiation. This is may be true in the case of radiation-induced necrosis. Thus, generally, these results confirm that, the relation between increase lysosomal enzyme and cell death.
In conclusion, in view to the recent studies on the effects of radiation on the biological structures, the current review throw up an important findings, the radiation induces release of ROS and hydrolytic enzymes which may cause tissue damage; involving of DNA. These changes may cause apoptosis and cell death.
REFERENCES
- Adams, J.D., S.K. Mukherjee, L.K. Klaidman, M.L. Chang and R. Yasharel, 1996. Apoptosis and oxidative stress in the aging brain. Ann. New York Acad. Sci., 786: 135-151.
PubMedDirect Link - Akagi, Y., K. Ito and S. Sawada, 1993. Radiation-induced apoptosis and necrosis in Molt-4 cells: A study of dose-effect relationships and their modification. Int. J. Radiat. Biol., 64: 47-56.
Direct Link - Alapetite, C., T. Wachter, E. Sage and E. Moustacchi, 1996. Use of the alkaline comet assay to detect DNA repair deficiencies in human fibroblasts exposed to UVC, UVB, UVA and gamma-rays. Int. J. Radiat. Biol., 69: 359-369.
Direct Link - Alien, J.W., P. Poorman-Allen, B.W. Collins and M.R. Sontag, 1994. Synaptonemal complex aberrations in the pseudoautosomal region of X,Y chromosomes in irradiated hamsters. Mutagenesis, 9: 259-267.
Direct Link - Backer, L.C., M.R. Sontag and J.W. Alien, 1991. Stage-specific damage to synaptonemal complexes and metaphase chromosomes induced by X-rays in male mouse germ cells. Radiat. Res., 125: 187-196.
PubMedDirect Link - Barham, S.S. and R.A. Walters, 1978. X-irradiation-induced nuclear lesions in cultured mammalian cells: An ultrastructural analysis. Radiat. Res., 76: 105-112.
Direct Link - Belpaeme, K., K. Cooreman and M. Kirsch-Voiders, 1998. Development and validation of the in vivo alkaline comet assay for detecting genomic damage in marine flatfish. Mutat Res., 15: 167-184.
Direct Link - Blakely, E.A. and A. Kronenberg, 1998. Heavy-ion radiobiology: New approaches to delineate mechanisms underlying enhanced biological effectiveness. Radiat. Res., 150: S126-S145.
Direct Link - Blank, K.R., M.S. Rudoltz, G.D. Kao, R.J. Muschel and W.G. McKenna, 1997. Review: The molecular regulation of apoptosis and implication for radiation oncology. Int. J. Radiat. Biol., 71: 455-466.
Direct Link - Brancolini, C., D. Lazarevic, J. Rodriguez and C. Schneider, 1997. Dismantling cell-cell contacts during apoptosis is coupled to a caspase-dependent proteolytic cleavage of beta-catenin. J. Cell Biol., 139: 759-771.
PubMedDirect Link - Brandes, D., K.W. Sloan, E. Anto and F. Bloedr, 1967. The effect of X-irradiation on the lysosomes of mouse mammary gland carcinoma. Cancer Res., 27: 731-746.
Direct Link - Brecher, S., 1977. Ultrastructural observations of X-ray induced chromatid gaps. Mutation Res., 42: 249-267.
PubMedDirect Link - Brinkley, B.R. and W.N. Hittelman, 1975. Ultrastructure of mammalian chromosome aberrations. Int. Rev. Cytol., 42: 49-101.
PubMedDirect Link - Bruge, F., L. Tiano, T. Cacciamani, F. Principi and G.P. Littarru, 2003. Effect of UV-C mediated oxidative stress in leukemia cell lines and its relation to ubiquinone content. Biofactors, 18: 51-63.
PubMedDirect Link - Burkart, W., T. Jung and G. Frasch, 1999. Damage pattern as a function of radiation quality and other factors. C. R. Acad. Sci. Paris, Sciences de la vie, Life Sci., 322: 89-101.
Direct Link - Cadet, J., T. Douki, J.P. Pouget and J.L. Ravanat, 2000. Singlet oxygen DNA damage products: Formation and measurement. Methods Enzymol., 319: 143-153.
PubMedDirect Link - Chan, W.H. and J.S. Yu, 2000. Inhibition of UV irradiation-induced oxidative stress and apoptotic biochemical changes in human epidermal carcinoma A431 cells by genistein. J. Cell Biochem., 78: 73-84.
PubMedDirect Link - Cheng, K.C., D.S. Cahill, H. Kasai, S. Nishimura and L.A. Loeb, 1992. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G-T and A-C substitutions. J. Biol. Chem., 267: 166-172.
PubMed - Clingen, P.H., C.F. Arlett, J. Cole, A.P. Waugh and J.E. Lowe et al., 1995. Correlation of UVC and UVB cytotoxicity with the induction of specific photoproducts in T-lymphocytes and fibroblasts from normal human donors. Photochem. Photobiol., 61: 163-170.
PubMedDirect Link - Conti, C.J., A.J.P. Klein-Szanto and J.L. Moreira, 1974. Effects of radiation on cultured BHK cells and their latent R particles. An electron microscope study. Archiv fur Geschwulstforschung, 43: 254-267.
Direct Link - Cramp, W.A., M.B. Yatvin and M. Harms-Ringdahl, 1994. Recent developments in the radiobiology of cellular membranes. Acta Oncol., 33: 945-952.
Direct Link - Daniniak, N. and B.J. Tann, 1995. Utility of biological membranes as indicators for radiation exposure: alterations in membrane structure and function overtime. Stem Cells, 13: 142-152.
PubMedDirect Link - Daya-Grosjean, L., N. Dumaz and A. Sarasin, 1995. The specificity of p53 mutation spectra in sunlight-induced human cancers. J. Photochem. Photobiol. B Biol., 28: 115-124.
CrossRefDirect Link - Desal, I.D., P.L. Sawant and A.L. Tappel, 1964. Peroxidative and radiation damage to isolated lysosomes. Biochim. Biophys. Acta, 86: 277-285.
PubMedDirect Link - DiPietro, R., E. Falcieri, M.A. Centurione, G. Mazzotti and R. Rana, 1994. Ultrastructural pattern of cell damage and death following gamma radiation exposure of murine erythroleukemia cells. Scanning Microscopy, 8: 667-673.
PubMedDirect Link - Drobetsky, E.A., J. Turcotte and A. Chateauneuf, 1995. A role for ultraviolet A in solar mutagenesis. Proc. Natl. Acad. Sci. USA., 92: 2350-2354.
Direct Link - Enninga, I.C., R.T. Groenendijk, A.R. Filon, A.A. van Zeeland and J.W. Simons, 1986. The wavelength dependence of U.V.-induced pyrimidine dimer formation, cell killing and mutation induction in human diploid skin fibroblasts. Carcinogenesis, 7: 1829-1836.
Direct Link - Eveno, E., F. Bourre, X. Quilliet, O. Chevallier-Lagente and L. Roza et al., 1995. Different removal of ultraviolet photoproducts in genetically related xeroderma pigmentosum and trichothiodystrophy diseases. Cancer Res., 55: 4325-4332.
Direct Link - Farber, J.L., 1982. Biology of disease; Membrane injury and calcium homeostasis in the pathogenesis of coagulative necrosis. Lab. Invest., 47: 114-123.
PubMedDirect Link - Firkle, T., V. Resl, J. Racek and V. Holecek, 2000. Antioxidants and protection of the skin against the effect of ultraviolet rays. Cas Lek Cesk., 139: 358-360.
PubMedDirect Link - Fox, J.C. and K.M. Prise, 1993. DNA lesions: Linear energy transfer and radiosensitive mutants. Br. J. Radiol. Suppl., 24: 48-52.
Direct Link - Freeman, S.E., H. Hacham, R.W. Gange, D.J. Maytum, J.C. Sutherland and B.M. Sutherland, 1989. Wavelength dependence of pyrimidine dimer formation in DNA of human skin irradiated in situ with ultraviolet light. Proc. Natl. Acad Sci. USA., 86: 5605-5609.
Direct Link - Fuks, Z., R.S. Persaud, A. Alfieri, M. McLoughlin and D. Ehleiter et al., 1994. Basic fibroblast growth factor protects endothelial cells against radiation-induced programmed cell death in vitro and in vivo. Cancer Res., 54: 2582-2590.
Direct Link - Gasperin, P., M. Gozy, O. Pauwels, J. Fruhling, P. VanHoutte, J.L. Pasteels and R. Kiss, 1992. Monitoring of radiotherapy-induced morphonuclear modifications in the MXT mouse mammary carcinoma by means of digital cell image analysis. Int. J. Radiat. Oncol. Biol., 22: 979-987.
PubMedDirect Link - Gobbel, G.T., M. Bellinzona, A.R. Vogt, N. Gupta, J.R Fike and P.H. Chan, 1998. Response of postmitotic neurons to X-irradiation: Implications for the role of DNA damage in neuronal apoptosis. J. Neurosci., 18: 147-155.
PubMedDirect Link - Goodhead, D.T., 1994. Initial events in the cellular effects of ionizing radiations: Clustered damage in DNA. Int. J. Radiat. Biol., 65: 7-17.
PubMedDirect Link - Gniadecki, R., B. Gajkowska, J. Bartosik, M. Hnasen and H.C. Wulf, 1998. Variable expression of apoptotic phenotype in keratinocytes treated with ultraviolet radiation, ceramide, or suspended in semisolid methyl-cellulose. Acta Dermato-Venereologica, 78: 248-257.
Direct Link - Guelman, L.R., L.M. Zieher and M.L. Fiszman, 1996. The effect of x-radiation on cerebellar granule cells grown in culture. Ganglioside GM1 neuro-protective activity. Neurochem. Int., 29: 521-527.
CrossRefDirect Link - Guelman, L.R., M.A. Zorrilla Zubilete, H. Rios, C.G. Di Toro, A.M. Dopico and L.M. Zieher, 2001. Motor, cytoarchitectural and biochemical assessment of pharmacological neuroprotection against CNS damage induced by neonatal ionizing radiation exposure. Brain Res. Protoc., 7: 203-210.
PubMedDirect Link - Guo, M., C. Chen, C. Vidair, S. Marino, W. Dewey and C.L. Clifton, 1997. Characterization of radiation induced apoptosis in rodent cell lines. Radiat. Res., 147: 295-303.
Direct Link - Haimovitz-Friedman, A., N. Balaban, M. McLoughlin, D. Ehleiter and J. Michaeli et al., 1994. Protein kinase C mediates basic fibroblast growth factor protection of endothelial cells against radiation-induced apoptosis. Cancer Res., 54: 2591-2597.
Direct Link - Hamberg, H., 1983. Cellular autophagocytosis induced by x-irradiation and vinblastine. On the origin of the segregating membranes. Acta Pathol. Microbiol. Scandinavica Section A Pathol., 91: 317-327.
PubMedDirect Link - Harris, J.W., 1967. Studies of irradiated lysosomes with particular reference to the action of calcium. Exp. Cell Res., 45: 487-489.
PubMedDirect Link - Hendry, J.H., 1997. West CML. Apoptosis and mitotic cell death: Their relative contributions to normal-tissue and tumor radiation response. Int. J. Radiat. Biol., 71: 709-719.
Direct Link - Humphrey, R. and B.R. Brinkley, 1969. Ultrastructural studies of radiation-induced chromosome damage. J. Cell Biol., 42: 745-753.
Direct Link - Ichihashi, M., M. Ueda, A. Budiyanto, T. Bito, M. Oka and M. Fukunaga et al., 2003. UV-induced skin damage. Toxicology, 189: 21-39.
CrossRefDirect Link - Ito, K. and S. Kawanishi, 1997. Site-specific DNA damage induced by UVA radiation in the presence of endogenous photo-sensitizer. Biol. Chem., 378: 1307-1312.
Direct Link - Johannisson, R., R. Mormel and B. Brandenburg, 1994. Synaptonemal complex damage in fetal mouse oocytes induced by ionizing irradiation. Mutation Res., 311: 319-328.
Direct Link - Jones, C.A., E. Huberman, M.L. Cunningham and M.J. Peak, 1987. Mutagenesis and cytotoxicity in human epithelial cells by far-and near-ultraviolet radiations: Action spectra. Radiat. Res., 110: 244-254.
PubMedDirect Link - Kadhim, M.A., S.A. Lorimore, K.M. Townsend, D.T. Godhead, V.J. Buckle and E.G. Wright, 1995. Radiation-induced genomic instability: Delayed cytogenetic aberrations and apoptosis in primary human bone marrow cells. Int. J. Radiat. Biol., 67: 287-293.
PubMedDirect Link - Kamat, J.P., K.K. Boloor, T.P. Devasagayam, B. Jayashree and P.C. Kesavan, 2000. Differential modification by caffeine of oxygen-dependent and independent effects of gamma-irradiation on rat liver mitochondria. Int. J. Radiat. Biol., 76: 1281-1288.
PubMedDirect Link - Kasai, H. and S. Nishimura, 1984. Hydroxylation of deoxyguanosine at the C-8 position by ascorbic acid and other reducing agents. Nucl. Acids Res., 12: 2137-2147.
PubMedDirect Link - Katz, D., D. Mazor, A. Dvilansky and N. Meyerstein, 1996. Effect of radiation on red cell membrane and intracellular oxidative defense systems. Free Radic. Res., 24: 199-204.
Direct Link - Kinashi, Y., Y. Sakurai, S. Masunage, M. Suzuki, K. Nagata and K. Ono, 2004. Ascorbic acid reduced mutagenicity at the HPRT locus in CHO cells against thermal neutron radiation. Applied Radiat. Isot., 61: 929-932.
CrossRefDirect Link - Koteles, G.J., 1979. New aspects of cell membrane radiobiology and their impact on radiation protection. Atomic Energy Rev., 17: 3-30.
PubMedDirect Link - Kondo, T., K. Takeuchi, Y. Doi, S. Yonemura, S. Nagaata and S. Tsukita, 1997. ERM (ezrin/radixin/ moesin)-based molecular mechanism of microvillar breakdown at an early stage of apoptosis. J. Cell Biol., 139: 749-758.
PubMedDirect Link - Kuo, S.S., A.H. Saad, A.C. Koong, G.M. Hahn and A.J. Giaccia, 1993. Potassium-channel activation in response to low doses of gamma-irradiation involves reactive oxygen intermediates in nonexcitatory cells. Proc. Natl. Acad. Sci. USA., 90: 908-912.
PubMedDirect Link - Kvam, E. and R.M. Tyrrell, 1997. Induction of oxidative DNA base damage in human skin cells by UV and near visible radiation. Carcinogenesis, 18: 2379-2384.
Direct Link - Lesnikov, V., M. Lesnikov and H.J. Deeg, 2001. Pro-apoptotic and anti-apoptotic effects of transferrin and transferrin-derived glycans on hematopoietic cells and lymphocytes. Exp. Hematol., 29: 477-489.
PubMedDirect Link - Lett, J.T., 1992. Damage to cellular DNA from particulate radiation, the efficacy of its processing and the radiosensitivity of mammalian cells. Emphasis on DNA strand breaks and chromatin break. Radiat. Environ. Biophys., 31: 257-277.
CrossRefDirect Link - Linnik, M.D., J.A. Miller, J. Sprinkle-Cavallo, P.J. Mason, F.Y. Thompson, L.R. Montgomery and K.K. Schroeder, 1995. Apoptotic DNA fragmentation in the rat cerebral cortex induced by permanent middle cerebral artery occlusion. Brain Res. Mol. Brain Res., 32: 116-124.
CrossRefDirect Link - Marrot, L., J.P. Belaid, J.R. Meunier, P. Perez and C. Agapakis-Causse, 1999. The human melanocyte as a particular target for UVA radiation and an endpoint for photoprotection assessment. Photochem. Photobiol., 69: 686-693.
PubMedDirect Link - Maes, A., L. Verschave, A. Arroyo, C. DeWagter and L. Vercruyssen, 1993. In vitro cytogenetic effects of 2450 MHZ waves on human peripheral blood lymphocytes. Bioelectromagnetics, 14: 495-501.
PubMedDirect Link - Maes, A., M. Collier, U. VanGorp, S. Vandoninck and L. Verschaeve, 1997. Cytogenetic effects of 935.2 MHz (GSM) microwaves alone and in combination with mitomycin C. Mutation Res., 393: 151-156.
PubMedDirect Link - Mansur, D.B., Y. Kataoka, D.J. Grdina and A.M. Diamond, 2001. Radiosensitivity of mammalian cell lines engineered to overexpress cytosolic glutathione peroxidase. Radiat. Res., 155: 536-542.
CrossRefDirect Link - Marrot, L., J.P. Belaid, J.R. Meunier, P. Perez and C. Agapakis-Causse, 1999. The human melanocyte as a particular target for UVA radiation and an endpoint for photoprotection assessment. Photochem. Photobiol., 69: 686-693.
PubMedDirect Link - Meloni, M. and J.F. Nicolay, 2003. Dynamic monitoring of glutathione redox status in UV-B irradiated reconstituted epidermis: Effect of antioxidant activity on skin homeostasis. Toxicol. In Vitro, 17: 609-613.
CrossRefDirect Link - Meng, H., T. Terado and H. Kimura, 1998. Apoptosis induced by X-rays and chemical agents in murine fibroblastic cell lines with a defect in repair of DNA double-strand breaks. Int. J. Radiation Biol., 73: 503-510.
Direct Link - Michel, P.P., N. Lambeng and M. Ruberg, 1999. Neuropharmacologic aspects of apoptosis: Significance for neurodegenerative diseases. Clin. Neuropharmacol., 22: 137-150.
Direct Link - Mills, J.C., N.L. Stone, N.L. Erhardt and R.N. Pittman, 1998. Apoptotic membrane blebbing is regulated by myosin light chain phosphorylation. J. Cell Biol., 140: 627-636.
Direct Link - Mitchell , D.L., J. Jen and J.E. Cleaver, 1991. Relative induction of cyclobutane dimers and cytosine photohydrates in DNA irradiated in vitro and in vivo with UV-C and UV-B light. Photochem. Photobiol., 54: 741-746.
PubMedDirect Link - Mitchell, D.L., 1988. The relative cytotoxicity of (6-4) photoproducts and cyclobutane dimers in mammalian cells. Photochem. Photobiol., 48: 51-57.
CrossRefDirect Link - Mironescu, S. and C. Dragomir, 1967. Nucleolar behaviour in regenerating liver of normal and whole-body-irradiated rats. Cancer Res., 27: 1819-1830.
PubMedDirect Link - Morales, A., A.E. Schwint and M.E. Itoiz, 1996. Nucleolar organiser regions in a model of cell hyperactivity and regression. Biocell, 20: 251-258.
Direct Link - Morgan, W.F., J.P. Day, M.I. Kaplan, E.M. McGhee and C.L. Limoli, 1996. Genomic instability induced by ionizing radiation. Radiat. Res., 146: 247-258.
PubMedDirect Link - Moustafa, Y.M., R.M. Moustafa, A. Belacy, S.H. Abou-El-Ela and F.M. Ali, 2001. Effects of acute exposure to the radiofrequency fields of cellular phones on plasma lipid peroxide and antioxidase activities in human erythrocytes. J. Pharm. Biomed. Anal., 26: 605-608.
PubMedDirect Link - Nakagawa, A., N. Kobayashi, T. Muramatsu, Y. Yamashina and T. Shirai et al., 1998. Three-dimensional visualization of ultraviolet-induced DNA damage and its repair in human cell nuclei. J. Invest. Dermatol., 110: 143-148.
PubMedDirect Link - Nakano, H. and K. Shinohara, 1994. X-ray-induced cell death: Apoptosis and necrosis. Radiat. Res., 140: 1-9.
PubMedDirect Link - Nguyen, T.D., F.X. Maquart and J.C. Monboisse, 2005. Ionizing radiations and collagen metabolism: from oxygen free radicals to radio-induced late fibrosis. Radiat. Phys. Chem., 72: 381-386.
Direct Link - Niiro, G.K. and T.M. Seed, 1988. SEM of canine chromosomes: Normal structure and the effects of whole-body irradiation. Scanning Microscopy, 2: 1593-1598.
PubMedDirect Link - Ojeda, F., H.A. Diehl and H. Folch, 1994. Radiation induced membrane changes and programmed cell death: Possible interrelationships. Scanning Microscopy, 8: 645-651.
Direct Link - Okunieff, P., M. Mester, J. Wang, T. Maddox and X. Gong et al., 1998. In vivo radioprotective effects of angiogenic growth factors on the small bowel at C3H mice. Radiat. Res., 150: 204-211.
Direct Link - Olive, P.L. and R.E. Durand, 1997. Apoptosis: An indicator of radiosensitivity in vitro. Int. J. Radiation Biol., 71: 695-707.
Direct Link - Orkisz, S. and H. Bartel, 1981. Morphology of nuclear ribonucleoprotein in the early period of liver regeneration after irradiation. Histochem. Cell Biol., 71: 629-634.
CrossRefDirect Link - Petit, P.X., H. Lecoeur, E. Zorn, C. Dauguet, B. Mignotte and M.L. Gougeon, 1995. Alterations in mitochondrial structure and function are early events of dexamethasone-induced thymocyte apoptosis. J. Cell Biol., 130: 157-167.
PubMedDirect Link - Piao, Y.J., K. Ogawa, K. Ono and M. Abe, 1983. The relationship between heterophagy and autophagy in the splenic macrophage of rats after γ-ray irradiation. Acta Histochem. Cytochem., 16: 353-367.
CrossRefDirect Link - Radford, I.R., 1999. Initiation of ionizing radiation-induced apoptosis: DNA damage-mediated or does ceramide have a role? Int. J. Radiat. Biol., 75: 521-528.
Direct Link - Rahman, Y.E., 1963. Effect of X-irradiation on the fragility of rat spleen lysosomes. Radiat. Res., 20: 741-750.
Direct Link - Rene, A.A., J.H. Darden and J.L. Parker, 1971. Radiation induced ultrastructural and biochemical changes in lysosomes. Lab. Inves., 25: 230-239.
PubMedDirect Link - Rosenstein, B.S. and D.L. Mitchell, 1987. Action spectra for the induction of pyrimidine (6‐4) pyrimidone photoproducts and cyclobutane pyrimidine dimers in normal human skin fibroblasts. Photochem. Photobiol., 45: 775-780.
CrossRefDirect Link - Roth, M.B., 1995. Spheres, coiled bodies and nuclear bodies. Curr. Opin. Cell Biol., 7: 325-328.
CrossRefDirect Link - Rugo, R.E. and R.H. Schiestl, 2004. Increases in oxidative stress in the progeny of X-irradiated cells. Radiat. Res., 162: 416-425.
Direct Link - Schwarz, A., S. Slander, M. Bemeburg, M. Bohm and D. Kulms et al., 2004. Interleukin-12 suppresses ultraviolet radiation-induced apoptosis by inducing DNA repair. Nat. Cell Biol., 4: 26-31.
CrossRefDirect Link - Shah, V.C. and P.K. Gadhia, 1979. Effect of sublethal dose of gamma-irradiation on lysosomal enzymes in tissue of pigeons. Radiat. Res., 20: 322-328.
PubMedDirect Link - Shimmura, S. and K. Tsubota, 1997. Ultraviolet B-induced mitochondrial dysfunction is associated with decreased cell detachment of corneal epithelial cells in vitro. Invest. Ophthalmol. Vision Sci., 38: 620-626.
PubMedDirect Link - Skog, S., V.P. Collins, B. Ivarson and B. Tribukait, 1983. Scanning and transmission electron microscopy following irradiation of Ehrlich ascites tumor cells. Relationships to cell cycle. Acta Radiol. Oncol., 22: 151-162.
PubMedDirect Link - Snyder, S.L. and S.K. Ekiund, 1978. Radiation-induced alteration in the distribution of lysosomal hydrolases in rat spleen homogenates. Radiat. Res., 75: 91-97.
Direct Link - Stapper, N.J., M. Stuschke, A. Sak and G. Stuben, 1995. Radiation-induced apoptosis in human sarcoma and glioma cell lines. Int. J. Cancer, 62: 58-62.
Direct Link - Stefani, S., S. Chandra and H. Tonaki, 1977. Ultrastructural events in the cytoplasmic death of lethally irradiated human lymphocytes. Int. J. Radiat. Biol., 31: 215-225.
Direct Link - Stohs, S.J. and D. Bagchi, 1995. Oxidative mechanisms in the toxicity of metal ions. Free Radic. Biol. Med., 18: 321-336.
CrossRefPubMedDirect Link - Somosy, Z., A. Pikli, T. Kubasova, G.J. Koteles and M. Bodo, 1985. Alteration of nuclear diameter of murine lymphocytes upon the effects of X-irradiation in vivo. Radiobiol. Radiother., 26: 305-310.
PubMedDirect Link - Somosy, Z., A. Takats, G. Bognar, A.L. Kovacs and A. Telbisz et al., 1996. X-irradiation-induced changes of the prelysosomal and lysosomal compartments and proteolysis in HT29 cells. Scann. Microscopy, 10: 1090-1091.
PubMedDirect Link - Farag, M.D.E.H., 1999. Effect of radiation and other processing methods on protein quality of sunflower meal. J. Sci. Food Agric., 79: 157-165.
Direct Link - Takahashi, H., Y. Hashimoto, N. Aoki, M. Kinouchi, A. Ishida-Yamamoto and H. Iizuka, 2000. Copper, zinc-superoxide dismutase protects from ultraviolet B-induced apoptosis of SV40-transformed human keratinocytes: The protection is associated with the increased levels of antioxidant enzymes. J. Dermatol. Sci., 23: 12-21.
PubMedDirect Link - Tanaka, T., 1979. Biochemical activity of nine lysosomal enzymes in T- and non-T-lymphocytes. Fed. Lett., 104: 161-264.
PubMedDirect Link - Tsoulou, E., C.A. Kalfas and E.G. Sideris, 2005. Conformational properties of DNA after exposure to gamma rays and neutrons. Radiat. Res., 163: 90-97.
CrossRefDirect Link - Vile, G.F. and R.M. Tyrrell, 1995. UVA radiation-induced oxidative damage to lipids and proteins in vitro and in human skin fibroblasts is dependent on iron and singlet oxygen. Free Radic. Biol. Med., 18: 721-730.
PubMedDirect Link - Vignais, P.V., 2002. The superoxide-generating NADPH oxidase: Structural aspects and activation mechanism. Cell. Mol. Life Sci., 59: 1428-1459.
CrossRefDirect Link - Waiters, R.L., 1992. Radiation-induced apoptosis in a murine T-cell hybridoma. Cancer Res., 52: 883-890.
Direct Link - Wang, S.Q., R. Setlow, M. Berwick, D. Polsky, A.A. Marghoob, A.W. Kopf and R.S. Bart, 2001. Ultraviolet A and melanoma: A review. J. Am. Acad. Dermatol., 44: 837-846.
CrossRefPubMedDirect Link - Wells, R.L. and A. Han, 1984. Action spectra for killing and mutation of Chinese hamster cells exposed to mid- and near-ultraviolet monochromatic light. Mutat Res., 129: 251-258.
PubMedDirect Link - Wenczl, E., G.P. Van der Schans, L. Roza, R.M. Kolb and A.J. Timmerman et al., 1998. (Pheo) Melanin photosensitizes UVA-induced DNA damage in cultured human melanocytes. J. Invest. Dermatol., 111: 678-682.
CrossRefDirect Link - Wyllie, A.H., J.F.R. Kerr and A.R. Currie, 1980. Cell death: The significance of apoptosis. Int. Rev. Cytol., 68: 251-306.
CrossRefPubMedDirect Link - Yamada, T. and H. Ohyama, 1988. Radiation-induced interphase death of rat thymocytes is internally programmed (apoptosis). Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med., 53: 65-75.
PubMedDirect Link - Zhao, X. and J.S. Taylor, 1996. Mutation spectra of TA, the major photoproduct of thymidylyl-(3`5`) deoxyadenosine, in Escherichia coli under SOS conditions. Nucl. Acid Res., 24: 1561-1565.
PubMedDirect Link - Zyss, R. and M. Kaszezynika, 1972. The lysosomal system of liver cells in rats following a radiation dose of 1000R to the whole body. Acta Med. Pol., 13: 177-192.
Direct Link