
Y. Baimark
Center of Excellence for Innovation in Chemistry, Department of Chemistry, Faculty of Science, Mahasarakham University, Mahasarakham 44150, Thailand
LiveDNA: 66.642
M. Srisa-ard
Center of Excellence for Innovation in Chemistry, Department of Chemistry, Faculty of Science, Mahasarakham University, Mahasarakham 44150, Thailand
A. Puntumchai
Center of Excellence for Innovation in Chemistry, Department of Chemistry, Faculty of Science, Mahasarakham University, Mahasarakham 44150, Thailand
Current Research in Chemistry
Year: 2010 | Volume: 2 | Issue: 1 | Page No.: 10-17







ABSTRACT
Terpolyesters of D, L-lactic acid (DLLA), Glycolic Acid (GA) and ε-caprolactone (CL), designed as PDLLGACL, were synthesized via direct polycondensation under reduced pressure at 160°C using tin(II) chloride dihydrate as a catalyst. The terpolyesters with different DLLA: GA: CL ratios were investigated. The appropriate polymerization time was 48 h that indicated from their percentage of yield and intrinsic viscosity. The number-average molecular weights from gel permeation chromatography curves of the polyesters were in the range of 6,600-7,900 g mol-1. The random monomer sequencing of co- and terpolyesters can be observed from the 1H- and 13C-NMR spectra. Glass transition temperatures of the polyesters were depended on their compositions and molecular weights.
PDF Abstract XML References Citation
How to cite this article
Y. Baimark, M. Srisa-ard and A. Puntumchai, 2010. Synthesis of Poly (D,L-lactic Acid-co-glycolic Acid-co-ε-caprolactone) Terpolyesters by Direct Polycondensation. Current Research in Chemistry, 2: 10-17.
DOI: 10.3923/crc.2010.10.17
URL: https://scialert.net/abstract/?doi=crc.2010.10.17
DOI: 10.3923/crc.2010.10.17
URL: https://scialert.net/abstract/?doi=crc.2010.10.17
INTRODUCTION
Homo-, co- and terpolyesters of D, L-lactic a cid, glycolic acid and ε-caprolactone have received much attention in the search for biodegradable polymers for potential use in biomedical and packaging applications. Each of the three homopolyesters: poly (DL-lactic acid), poly (glycolic acid) and poly (ε-caprolactone) is biodegradable via a simple, non-enzymatic hydrolysis mechanism (Srisa-ard et al., 2001). The hydrolysis products are non-toxic. However, the properties of these homopolyesters invariably only partially match the property requirements of the application such as biodegradation rate and mechanical properties. The co- and terpolyesters of these monomers have all been reported either as random or block co- and terpolymers (Florczak et al., 2007; Yaoming et al., 2007). Random copolymers with different compositions and types of monomer units give a range of materials with different mechanical and biodegradative properties (Saha and Tsuji, 2006).
Usually, terpolyesters of these monomers were synthesized by ring-opening polymerization. But this technique involves too many steps and is not cost-effective (Yaoming et al., 2007). Direct polycondensation for preparing polyesters is a faster and cheaper method than the ring-opening polymerization. Many works have been reported on polycondensation of homopolyesters of L-lactic and glycolic acids (Takahashi et al., 2000; Moon et al., 2001; Chen et al., 2006; Takasu et al., 2006; Achmad et al., 2009). The synthesis of copolyesters by polycondensation has been scarcely published (Yaoming et al., 2007; Hirata and Kimura, 2008).
In this study, we report the direct polycondensation of terpolyesters of D, L-lactic acid, glycolic acid and ε-caprolactone. The optimum polymerization time was determined. The structures and thermal properties of terpolyesters with varying compositions have been investigated and their structure-property relationships are discussed.
MATERIALS AND METHODS
This research was conducted on October 2008-August 2009 at Mahasarakham University, Mahasarakham, Thailand.
Excess water in DL-lactic acid (DLLA) aqueous solution (90 wt.%, Fluka, Switzerland) was distilled in the absence of any catalyst under reduced pressure at 150°C before use (Chen et al., 2006). The ε-caprolactone (CL, 99%, Acros Organics, USA) was purified via drying with CaH2 followed by distillation under reduced pressure before being stored over molecular sieves in a refrigerator. Glycolic acid (GA, 99%, Acros Organics, USA) and tin (II) chloride dehydrate (SnCl2.2H2O, 98%, Carlo Erba, USA) were used without further purification. Other reagents were of analytical grade.
Synthesis of Terpolyesters
Poly(D,L-lactic acid-co-glycolic acid-co-ε-caprolactone) terpolyesters (PDLLGACL) was synthesized by polycondensation in a round-bottom flask equipped with a magnetic stirrer, which had been dried in an oven at 150°C for 24 h before use. Appropriate amounts of monomers and SnCl2.2H2O were charged into the reactor. Table 1 shows DLLA:GA:CL feed ratios of 100: 0: 0, 90: 10: 0 and 90: 5: 5 by weight for the synthesis of PDLLA, PDLLGA and PDLLGACL, respectively. The polymerization procedure was described by Yaoming et al. (2007) with some modification. The round-bottom flask was immersed in an oil bath maintained at 160°C under reduced pressure for 48 h. The SnCl2.2H2O concentration was kept constant at 0.5 wt. %.
| Table 1: | Characteristics of the terpolyesters |
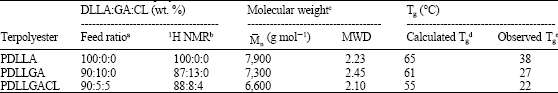 | |
| aCalculated from monomer feed ratios, bCalculated from 1H-NMR spectra, cObtained from GPC curves, | |
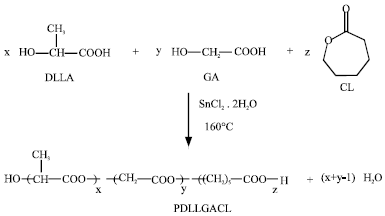 | |
| Scheme 1: | Polymerization reaction of PDLLGACL |
The DLLA, GA and CL monomers were synthesized to obtain terpolymer using SnCl2.2H2O as a catalyst at 160°C, as shown in Scheme 1. The as-polymerized terpolyesters were purified by dissolving in chloroform before being precipitated in cold n-hexane. Finally, they were dried to constant weight in a vacuum oven at room temperature before characterization.
Characterization of Terpolyesters
The intrinsic viscosity (η) of terpolyesters were determined from flow-time measurements on a diluted series of solutions in chloroform (CHCl3) as solvent at 30°C using viscometer. Terpolymer compositions and microstructures of the polyesters were characterized by 1H-NMR using a Bruker Avance DPX 300 1H-NMR Spectrometer. Spectra were obtained from copolymer solutions in deuterated chloroform using tetramethysilane as internal reference. Number-average molecular weights,![]() and molecular weight distributions, MWD, were determined by Gel Permeation Chromatography (GPC) using a Waters 717 plus Autosampler GPC equipped with an Ultrastyragel® column operating at 40°C and employing universal calibration. Tetrahydrofuran was used as the solvent at a flow rate of 1.0 mL min-1. Thermal analysis was carried out by means of Differential Scanning Calorimetry (DSC) using a Perkin-Elmer DSC Pyris Diamond. For DSC analysis, terpolymer samples weighing 5-10 mg were heated at 10°C min-1 under a helium atmosphere in order to observe their glass transition temperatures (Tg) from their second heating scans. For the second heating scans, the terpolyesters were first heated to 100°C before fast cooling (quenching) according to the DSC instrument’s own default cooling mode before the second run.
and molecular weight distributions, MWD, were determined by Gel Permeation Chromatography (GPC) using a Waters 717 plus Autosampler GPC equipped with an Ultrastyragel® column operating at 40°C and employing universal calibration. Tetrahydrofuran was used as the solvent at a flow rate of 1.0 mL min-1. Thermal analysis was carried out by means of Differential Scanning Calorimetry (DSC) using a Perkin-Elmer DSC Pyris Diamond. For DSC analysis, terpolymer samples weighing 5-10 mg were heated at 10°C min-1 under a helium atmosphere in order to observe their glass transition temperatures (Tg) from their second heating scans. For the second heating scans, the terpolyesters were first heated to 100°C before fast cooling (quenching) according to the DSC instrument’s own default cooling mode before the second run.
RESULTS
Figure 1 shows percentage yields with different polymerization times of polyesters. The percentage yields increased with polymerization time increased until 48 h. It can be seen that the percentage yields of PDLLA and PDLLGA at the same polymerization time are similar. When CL monomer was copolymerized, the percentage yield slightly decreased. The influence of polymerization time on the intrinsic viscosity, as shown in Fig. 2 indicated that the intrinsic viscosity increased as the increasing of polymerization time. When the polymerization time was increased to 72 h, the intrinsic viscosity was decreased. The results suggested that the appropriate polymerization time at 160°C was 48 h for all polyesters.
The compositions of co-and terpolyesters were determined from their 1H-NMR spectra by using the peak areas corresponding to the DLL methine protons at δ = 5.0-5.3 ppm, the G methylene protons at δ = 4.5-4.9 ppm and the CL ε-methylene protons at δ = 3.9-4.2 ppm. The 1H-NMR spectra of the polyesters are shown in Fig. 3a-c and the calculated compositions of DLLA/GA/CL are given in Table 1.
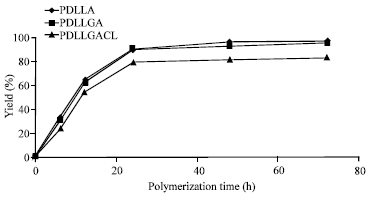 | |
| Fig. 1: | Relationship between percentage yields and polymerization time |
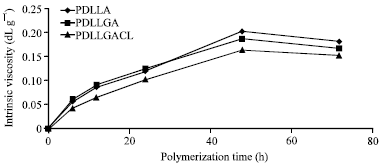 | |
| Fig. 2: | Relationship between intrinsic viscosity and polymerization time |
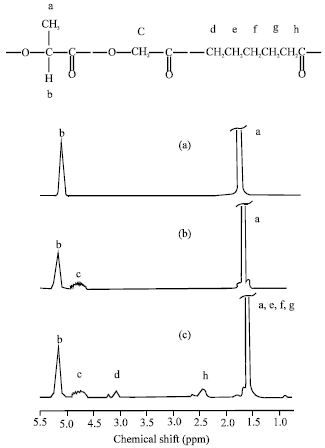 | |
| Fig. 3: | 1H-NMR spectra of (a) PDLLA, (b) PDLLGA and (c) PDLLGACL (peak assignments as shown) |
As would be expected, the terpolymer compositions are similar to the monomer feed ratios.
Molecular weight characterization was carried out by means of GPC. The molecular weight characteristics are also reported in Table 1. It was found that the ![]() of PDLLGA and PDLLGACL were lower than the PDLLA. All polyesters gave similar unimodal GPC molecular weight distributions.
of PDLLGA and PDLLGACL were lower than the PDLLA. All polyesters gave similar unimodal GPC molecular weight distributions.
The chain microstructures of polyesters are reflected in the fine structures of the 1H-NMR spectra. The appearance of multiple resonances for the same proton can be attributed to the presence of different monomer sequences and therefore slightly different chemical environments in the co- and terpolymer chains.
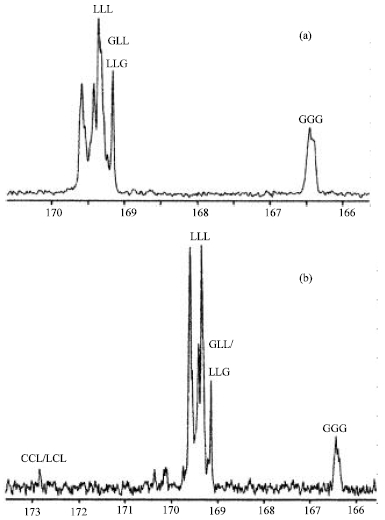 | |
| Fig. 4: | Expanded carbonyl regions of 13C-NMR spectra of (a) PDLLGA and (b) PDLLGACL |
The CH2 protons in the G units and α-CH2 and ε-CH2 protons in the CL units are seen to be particularly sensitive to this. The band at 4.8 ppm corresponding to the CH2 protons of the G units shows different resonance lines indicating various sequences of lactyl (or lactic acid) units and glycolyl (or glycolic acid) units (Baimark et al., 2007). The bands at 2.4 and 4.1 ppm corresponding to the α-CH2 and ε-CH2 protons in the CL units, respectively are split into two quite distinct triplets adjacent to one another. Monomer sequencing was characterized from the 13C-NMR spectra, specially from the expanded carbonyl carbon (C = O) region from δ = 166-173 ppm, as shown in Fig. 4 for PDLLGA and PDLLGACL. The various peaks can be assigned to various carbonyl carbons of the middle units of triad sequences, as also labelled in Fig. 4a and b (L = lactyl unit, G = glycolyl unit and C = ε-caprolactone unit). The expanded carbonyl region of 13C-NMR spectrum of PDLLA showed only LLL triad peak. The various triad peaks, excepted LLL and GGG peaks in Fig. 4 indicated the random monomer sequencing of the co-and terpolyesters (Baimark and Molloy, 2005; Baimark et al., 2007).
Thermal analysis of the terpolyesters was carried out by means of DSC. The DSC curves of the terpolyesters each exhibited a single Tg over the range of 20-40°C, as shown in Fig. 5 and also summarized in Table 1. The Tgs of the PDLLA, PDLLGA and PDLLGACL are 38, 27 and 22°C, respectively. The Tg of PDLLA decreased by incorporating glycolide and ε-caprolactone units into the polyester chain.
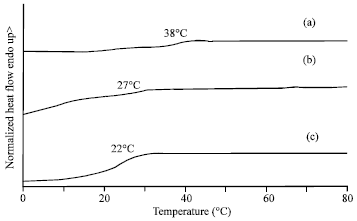 | |
| Fig. 5: | DSC thermograms of (a) PDLLA, (b) PDLLGA and (c) PDLLGACL |
DISCUSSION
The tin (II) chloride dehydrate has been used as a catalyst for polycondensation of poly(ε-caprolactone-co-lactic acid) (Yaoming et al., 2007). It is reported that the tin (II) chloride dehydrate with concentration of 0.5% by weight showed good dispersibility and dissolubility during polymerization and gave high polyester molecular weight. From Figure 1, the percentage yields of all polyesters did not increase after polymerization time of 48 h. It suggested that the highest %conversion was obtained at 48 h. The percentage yield was nearly 100% indicating that the synthesis reactions proceeded to near-quantitative conversion. Meanwhile, the intrinsic viscosities of all polyesters were decreased when polymerization time was higher than 48 h. This may be interpreted in term of degradative side reactions catalyzed by tin (II) chloride dehydrate taking place. Then, the polyesters prepared using polymerization time at 48 h were chosen for characterization of molecular weight, chain microstructure and thermal property.
The ![]() of all polyesters obtained from GPC analysis were directly related to their intrinsic viscosities, as shown in Table 1. The molecular weights of PDLLA and PDLLGA were slightly higher than PDLLGACL. This may be due to the percentage yield of PDLLGACL was lower than those of PDLLA and PDLLGA (Fig. 1).
of all polyesters obtained from GPC analysis were directly related to their intrinsic viscosities, as shown in Table 1. The molecular weights of PDLLA and PDLLGA were slightly higher than PDLLGACL. This may be due to the percentage yield of PDLLGACL was lower than those of PDLLA and PDLLGA (Fig. 1).
The CH2 peaks of G and CL units in 1H-NMR spectra can be used to determine comonomer sequencing (Baimark et al., 2007). The multiple peak split of CH2 protons of the G units (Fig. 3b) attributed to random sequencing of DLL and G units. While the multiple peak split of α-CH2 and ε-CH2 protons of the CL units (Fig. 3c) designed as random sequencing of DLL and CL units. The randomization of monomer sequencing of co- and terpolyesters can be confirmed from expanded carbonyl region of 13C-NMR spectra, as shown in Fig. 4. The L, G and C in the peak labeling are haft-lactide, haft-glycolide and ε-caprolactone, respectively. These triad sequencing occurred by tranesterification reaction during polycondensation. The both 1H- and 13C-NMR results supported the random character of the co- and terpolyesters.
For DSC analysis, the Tg of polyesters are in order of PDLLA>PDLLGA>PDLLGACL because the Tg of homopolyester of PDLLA>PDLLGA>PCL. Each experimentally observed Tg of the polyesters is comparable with the weight-averaged value calculated from the Fox Eq. 1 for a random terpolymer.
| (1) |
where, wDLLA, wGA and wCL are the respective weight fractions of the DLLA, GA and CL units, as calculated from the corresponding mole fractions from 1H-NMR. TgPDLLA (338 K), TgPGA (308 K) and TgPCL (213 K) are the respective Tg (K) values of the PDLLA, PGA and PCL homopolymers, as obtained from the reference literature (Baimark et al., 2007).
The calculated Tg values of the terpolyesters from the Fox Equation are summarized in Table 1. It is found that the Tgs from the DSC curves are generally lower than the calculated Tg values from the Fox Equation. This may be due to these polyesters are low molecular weight polymers. The results suggested that the Tg of polymer also directly related to the molecular weight of ![]() polymer. The Tg of poly(L-lactide-co-glycolide-co-ε-caprolactone) terpolyester obtained from DSC curve (35°C) was closer to the Tg calculated from the Fox equation (38°C) because of its was approximately 105 g mol-1 (Srisa-ard et al., 2001).
polymer. The Tg of poly(L-lactide-co-glycolide-co-ε-caprolactone) terpolyester obtained from DSC curve (35°C) was closer to the Tg calculated from the Fox equation (38°C) because of its was approximately 105 g mol-1 (Srisa-ard et al., 2001).
Finally, it should be noted that the polycondensation method is a simple and low-cost method for synthesizing terpolyesters compared with ring-opening polymerization. It is possible in large-scale production for use as biodegradable polyesters in drug delivery applications (Zhao et al., 2004).
CONCLUSION
The PDLLGACL with different chemical compositions were successfully synthesized by direct polycondensation under reduced pressure at 160°C using tin (II) chloride dehydrate as the catalyst. The polymerization time at 48 h gave the highest molecular weight polyesters prepared by this method. The tin (II) chloride dehydrate shows superior to the conversional systems in regard to randomization of the monomers in resultant co- and terpolyesters. The random character of polyesters can be analyzed from their 1H- and 13C-NMR spectra. The Tgs of polyesters depended on their compositions and molecular weights. It is expected that these low molecular weight terpolyesters will find potential applications in biomedical and pharmaceutical fields.
ACKNOWLEDGMENTS
This study was supported by the National Research of Council of Thailand, the Research Development and Support Unit, Mahasarakham University (fiscal year 2009) and the Center of Excellence for Innovation in Chemistry (PERCH-CIC), Commission on Higher Education, Ministry of Education, Thailand.
REFERENCES
- Achmad, F., K. Yamane, S. Quan and T. Kokugan, 2009. Synthesis of polylactic acid by direct polycondensation under vacuum without catalysts, solvents and initiators. Chem. Eng. J., 151: 342-350.
CrossRef - Baimark, Y. and R. Molloy, 2005. Synthesis and characterization of poly(L-lactide-co-ε-caprolactone) (B)-poly(L-lactide) (A) ABA block copolymers. Polym. Adv. Tech., 16: 332-337.
CrossRefDirect Link - Chen, G.X., H.S. Kim, E.S. Kim and J.S. Yoon, 2006. Synthesis of high-molecular-weight poly(L-lactic acid) through the direct condensation polymerization of L-lactic acid in bulk state. Eur. Polym. J., 42: 468-472.
CrossRefDirect Link - Florczak, M., J. Libiszowski, J. Mosnacek, A. Duda and S. Penczek, 2007. L,L-lactide and -caprolactone block copolymers by a poly(L,L-lactide) block first route. Macromol. Rapid Commun., 28: 1385-1391.
CrossRef - Hirata, M. and Y. Kimura, 2008. Thermomechanical properties of stereoblock poly(lactic acid)s with different PLLA/PDLA block compositions. Polymer, 49: 2656-2661.
CrossRefDirect Link - Srisa-ard, M., R. Molloy, N. Molloy, J. Siripitayananon and M. Sriyai, 2001. Synthesis and characterization of a random terpolymer of L-lactide, -caprolactone and glycolide. Polym. Int., 50: 891-896.
CrossRef - Moon, S.I., C.W. Lee, I. Taniguchi, M. Miyamoto and Y. Kimura, 2001. Melt/solid polycondensation of L-lactic acid: An alternative route to poly(L-lactic acid) with high molecular weight. Polymer, 42: 5059-5062.
Direct Link - Saha, S.K. and H. Tsuji, 2006. Hydrolytic degradation of amorphous films of L-lactide copolymers with glycolide and D-lactide. Macromol. Mater. Eng., 291: 357-368.
CrossRef - Takahashi, K., I. Taniguchi, M. Miyamoto and Y. Kimura, 2000. Melt/solid polycondensation of glycolic acid to obtain high-molecular weight poly(glycolic acid). Polymer, 41: 8725-8728.
CrossRef - Takasu, A., Y. Narukawa and T. Hirabayashi, 2006. Direct dehydration polycondensation of lactic acid catalyzed by water-stable lewis acids. J. Polym. Sci. Part A: Polym. Chem., 44: 5247-5253.
CrossRef - Yaoming, Z., Z. Ling and W. Zhaoyang, 2007. Direct melting polycondensation and characterization of poly(-caprolactone-co-lactic acid). Frontiers Chem. China, 2: 178-182.
CrossRefDirect Link - Zhao, Y.M., Z.Y. Wang, J. Wang, H.Z. Mai B. Yan and F. Yang, 2004. Direct synthesis of poly(D,L-lactic acid) by melt polycondensation and its application in drug delivery. J. Applied Polym. Sci., 91: 2143-2150.
CrossRef