
Wu-Yi Liu
Department of Science and Technology, Fuyang Normal College, China
Biotechnology
Year: 2014 | Volume: 13 | Issue: 4 | Page No.: 190-195







ABSTRACT
Polymerase Chain Reaction (PCR) is a fundamental in vitro technique to molecular biology in practice. However, the utility of PCR and PCR-based methods is dependent on the unique of identifying and designing and efficient primer sequences. In silico PCR can thus assist in selection of newly designed primers, identify potential mismatches in the primer binding sites and greatly avoid the amplification issues of unwanted amplicons before any real experiments in practice. The present study was aimed to design and select novel primers and validate reported primers for multi-exon genes in practical investigation procedure on the Single Nucleotide Polymorphism (SNP) of PRKAG2 and PRKAG3 gene loci in Chinese native chicken populations applying in silico PCR analysis strategy. After careful experimental operation and comparison, it was found that results of in silico PCR analysis and the real experimental PCR amplifications were strikingly in resemblance.
PDF Abstract XML References Citation
Received: March 23, 2014;
Accepted: July 04, 2014;
Published: August 20, 2014
How to cite this article
Wu-Yi Liu, 2014. Application of in silico PCR Strategy for Primer Design and Selection
of Chicken AMPK Gamma Subunit Gene Loci. Biotechnology, 13: 190-195.
DOI: 10.3923/biotech.2014.190.195
URL: https://scialert.net/abstract/?doi=biotech.2014.190.195
DOI: 10.3923/biotech.2014.190.195
URL: https://scialert.net/abstract/?doi=biotech.2014.190.195
INTRODUCTION
Polymerase Chain Reaction (PCR) is a fundamental technique for the amplification of target gene sequences or DNA segments in vitro. The PCR technique was first described by Saiki et al. (1985) and perfected by Saiki et al. (1988) and Mullis et al. (1986, 1992). Since, the introduction of optimized PCR adopting heat-resistant and thermo-tolerant Taq polymerase in 1988, the PCT procedure was greatly simplified and this enabled the automation process of PCR reactions (Saiki et al., 1988). Later, automated PCR has been extensively used in gene discovery, molecular diagnosis and genetic DNA typing, etc. This technique generally can produce a million copies from a single template through no less than 20 cycles of template denaturing, primer annealing and product extension. The sensitivity of PCR originates from its exponential amplification while PCR specificity is determined by a pair of oligonucleotide DNA sequences, known as primers (Bartlett and Stirling, 2003). The PCR technique can be used to identify or isolate a specific DNA fragment from a complex genome. Thus, PCR and PCR-based methods are regarded as fundamental to molecular biology and the most important practical molecular techniques for the modern laboratories. However, the utility of the method is dependent on identifying unique primer sequences and designing PCR-efficient primers. Primer design is thus a critical step in all types of PCR methods to ensure specific and efficient amplification of a target sequence and therefore many researchers were committed to develop efficient software suites for primer designing and selection and verification (Rubin and Levy, 1996; Nishigaki et al., 2000; Yuryev et al., 2002; Kent et al., 2002; Lexa and Valle, 2003; Marshall, 2004; Boutros and Okey, 2004; Gadberry et al., 2005; Cao et al., 2005; Housley et al., 2006; Aranyi et al., 2006; Aranyi and Tusnady, 2007; Fredslund and Lange, 2007; Bekaert and Teeling, 2008; Kalendar et al., 2011; Kalendar et al., 2014).
In practice, PCR primers are usually 18-30 nucleotides in length giving them high specificity. However, this theoretical prediction may not always be true in diverse and complicated biological genomes. The last 10-12 bases at the 3’ end of primers are important for binding stability; single mismatches can reduce PCR efficiency, the effect increasing with proximity to the 3’ end. Non-specific amplification with unexpected amplicons appears frequently and the “Trial and error testing” is laborious and time consuming. Moreover, PCR might be misleading in DNA diagnosis if there is an allele drop due to polymorphism-induced mismatches in primer binding sites. Publication errors in primer sequences can lead to amplification failure or even wrong amplification of an unwanted target. In silico PCR refers to a virtual PCR executed by a computer and programs with the input of a pair or a batch of primers against an intended genome and/or sequence databases. With the development of sequencing technology and rapid cost reduction, many genomes have been sequenced and annotated in databases. Such a wealth of genomic information makes in silico PCR analysis possible. In silico PCR aims to test PCR specificity and identify the mismatches in primer binding sites due to known Single Nucleotide Polymorphisms (SNPs) and/or unwanted amplicons from a pseudogene or a homologous gene. In silico PCR analysis is an efficient complementary method to ensure primer specificity for an extensive range of PCR applications for gene discovery, molecular diagnosis and genetic DNA typing. In silico PCR method can assist in the selection of newly designed primers, identify potential mismatch issues in the primer binding sites due to known SNPs and avoid the amplification issues of unwanted amplicons before any real experiments. For instance, in silico PCR analysis can assist in the selection of newly designed primers, especially of multi-exon genes, for the evaluation of DNA barcodes for fungi and revealed potential PCR biases (Bellemain et al., 2010; Ficetola et al., 2010) to avoid potential problems before primer synthesis and experiments. Moreover, in silico PCR analysis is also useful to validate published primers before being blindly adopted, such as the evaluation of reported primers coded for the 16S rRNA (16S rRNA) subunit and flagellin B (FlaB) gene loci (Noda et al., 2013).
AMP-activated protein kinase (AMPK) is a heterotrimer consisting of an alpha catalytic subunit and non-catalytic beta and gamma subunits. The AMPK is an important energy-sensing enzyme that monitors cellular energy status. In response to cellular metabolic stresses, AMPK is responsible for the regulation of fatty acid synthesis by phosphorylation and regulates cholesterol synthesis via., phosphorylation and hormone-sensitive lipase too. All the subunits of AMPK are required to show significant kinase activity and AMPK plays an important role in the regulation of glucose and lipid metabolism in skeletal muscle tissues. The protein encoded by PRKAG (AMPK gamma) gene loci is a regulatory subunit of AMPK and mainly appears and expressed in skeletal muscle tissues. There are totally three gamma subunit isoforms of AMPK, i.e., γ-1, γ-2 and γ-3 subunit gene loci encoded by multiple exons denoted as PRKAG1, PRKAG2 and PRKAG3. There is a major significance for us to understand the function and regulatory mechanism of AMPK and to screen Single-Nucleotide Polymorphisms (SNPs) in AMPK gamma gene loci and their corresponding biological effects. This study was especially aimed to design and select efficiently new primers for these multi-exon genes for subsequent practical investigation of the Single Nucleotide Polymorphism (SNP) of PRKAG2 and PRKAG3 gene loci in Chinese native chicken breeds.
MATERIALS AND METHODS
A computer and the internet access are required for in silico PCR analysis. The web-based in silico PCR software suites or website tool utilized and discussed in this study is mainly the “in silico PCR” from UCSC Genome Browser (http://genome.cse.ucsc.edu/; University of California, Santa Cruz; Kent et al., 2002; Karolchik et al., 2014). The primer design and in silico PCR analysis were based on the multi-exon nucleotide sequences of chicken PRKAG gene loci in the databases of GenBank (Fig. 1), i.e., DQ212708.1, DQ212709.1, DQ212710.1, DQ212711.1, NM_001278142.1, NM_001278143.1, NM_001030965.2 and NC_006089 (for PRKAG2); DQ079814.2, DQ079815.2, NM_001031258.2 and NC_006094 (for PRKAG3) (Fig. 1). Some primers were designed with off-line primer design softwares, such as Oligo and Primer Premier and artificially improvements while the design of other PRKAG3 primers was referred from Zhao et al. (2006).
DNA sampling: The chicken muscle samples were taken from individuals of the same breed in the slaughter house, fed using standard commercial protocol with the same diet and slaughtered under the same environment and management rearing.
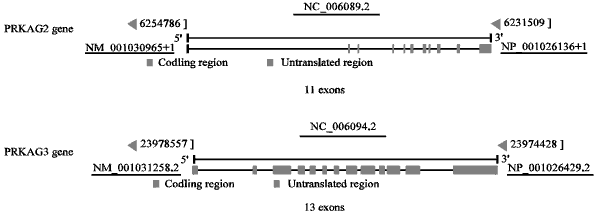 | |
| Fig. 1: | Multi-exon nucleotide sequences of chicken PRKAG2 and PRKAG3 gene loci |
Within 10 min after the animals were euthanized, chicken chest muscle tissue samples were fetched and genomic DNA was extracted using the standard phenol-chloroform or alkaline protocols (Sambrook, 2001), stored at -20°C for subsequent usage.
Methods: In silico PCR amplifications were executed for the examination and pre-analysis of objective primers designed before practice. The in silico PCR program of UCSC Genome Browser is faster than other similar programs since an indexing strategy is used in the program.
In silico PCR analysis and/or evaluation of reported primers: The in silico PCR program of UCSC Genome Browser can be initiated by three required steps as followed (Yu and Zhang, 2011):
| • | Primer sequences to be tested: Forward and reverse primers could be typed into two text boxes as indicated in the UCSC Genome Browser. If both primer sequences are originated from the same strands, the box of Flip Reverse Primer should be ticked. The program will automatically take the reverse and complement sequences before in silico PCR analysis. The default minimal length is 15 nucleotides for any primers |
| • | Selection of target genomes: There are two dropdown menus of “Genome” and “Assembly.” One can select a target genome from diverse model organisms listed and a particular version of the genome assembly. Additional target options of the genome assembly and UCSC genes are available only for human and mouse genomes (Kent et al., 2002; Karolchik et al., 2003, 2014). The user can choose in silico PCR template as either genomic DNAs or cDNAs (for expressed transcripts) in the human and mouse genomes |
| • | PCR parameters setting: The box of Max Product Size in the UCSC Genome Browser has the default value of 4000 bp which allows users to define the maximal size of the expected in silico PCR amplicons. Any amplified amplicon larger than a defined value will be filtered out. Both “Min Perfect Match” and “Min Good Match” define the stringency of primers. The former refers to minimal number of nucleotides (no less than 15) on the 3’ ends of primers that must exactly match the template target. “Min Good Match” is only relevant to the nucleotides beyond the number defined by “Min Perfect Match,” among which they should have two-thirds matching to the PCR template target |
It should be noted that both “Min Perfect Match” and “Min Good Match” are not critical and the default values can be used without further modification. Click on the summit button after all inputs are completed. A typical display will be shown in the “Result page”. During the evaluation, partial sequences of target gene were also obtained and validated from GenBank (http://www.ncbi.nlm.nih.gov/) and aligned using Clustal (www.ebi.ac.uk/ clustalw/). The specificity of primers was verified by BLAST program (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The dimer formation was obtained using OPERON (http://www.operon.com/technical/toolkit.aspx). Other parameters like the melting and annealing temperature, primer length, GC content and 3’-extreme stability were evaluated using the BioEdit software (www.bioedit.com).
Real PCR amplification and visualization: Real PCR amplification program was carried out after primer synthesis in a total volume of 25 μL. The PCR amplification mixture contained 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 0.25 mM of dNTP each, 0.01 mg Bovine Serum Albumin (BSA), 50 ng of each primer, 0.05 units of Taq polymerase and about 50 ng of genomic DNA. The graded PCR amplification programs were designed with an initial denaturing step at 94°C for 5 min, followed by 35 cycles at 94°C for 30 sec, annealing at 60±9°C for 30 sec and 72°C for 45 sec and final extention step at 72°C for 10 min with a final hold at 4°C. The PCR products were then separated by electrophoresis on 1.5% agarose gel for 25-30 min at 120 V and stained with EB (ethidium bromide) for visualization under UV light.
RESULTS AND DISCUSSION
Validation and selection of primers: To amplify the partial sequences of multiple-exons of PRKAG2 and PRKAG3, we initially designed more than 30 pair primers according to the gene sequences provided in the database of GenBank. The results of in silico PCR analysis and real experimental PCR amplification were strikingly in resemblance. All the newly designed pair primers for PRKAG2 and PRKAG3 were verified and selected in subsequent experiments. The results of in silico PCR analysis and real PCR amplifications were strikingly in resemblance (Table 1 and 2). For simplification, only the primers and real PCR amplifications for PRKAG3 referred from Zhao et al. (2006) were further analyzed and discussed (Table 1). The results of in silico PCR amplification, analysis and selection of PRKAG3 primers suitable for subsequent PCR-based experiments were showed in Table 1 and 2.
| Table 1: | In silico PCR analysis of PRKAG3 primers from Zhao et al. (2006) |
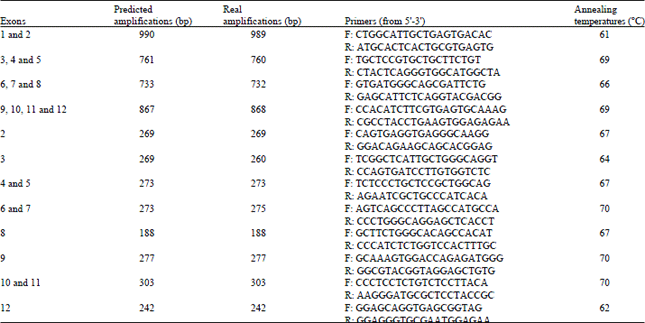 | |
| Table 2: | In silico PCR analysis of PRKAG2 and PRKAG3 primers |
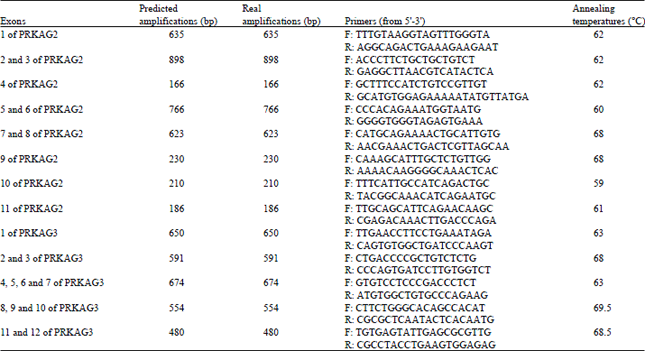 | |
PCR amplifications: Real PCR amplification products were discerned by 1.5% agarose gel electrophoresis detections, with bright target fragments for subsequent SNP analysis (Fig. 2).
In silico PCR analysis could be useful and efficient in the identification of potential problems before any real experiments or validate published primers. As seen from Table 1, Fig. 1 and 2, “No matches” can result from many underlying causes besides the wrong primer sequences. For example, an exon of PRKAG3 gene loci over hanging primer won’t find any target in the chicken genome assembly database rather than in a gene transcript. Another successful application example of in silico PCR analysis was the rapid sex diagnosis of genotypes of multi-exon chromo-helicase DNA-binding (CHD) gene loci in both avian Z and W chromosomes (Liu et al., 2010).
 | |
| Fig. 2: | Real PCR amplification photos of chicken PRKAG3 gene loci discerned by 1.5% agarose gel electrophoresis detections. M: Marker |
In practical in silico PCR analysis, it is possible that a primer has been designed on a polymorphic region that is absent from the database sequence. A well-known shortcoming of this method is that deletion fragments larger than the amplicon sizes can be ignored due to the diploid nature of the domestic animal genomes. In this case, only hemizygous PCR amplification of the undeleted fragment may occur. Therefore, it is quite useful to know how many SNPs are expected in an amplicon through an in silico analysis (Yu and Zhang, 2011) which can exclude the potential miss-diagnosis due to the failed primers’ binding caused by known polymorphisms.
ACKNOWLEDGMENTS
We are grateful to the anonymous reviewers for their suggestions. The partial financial support of the following programs is also greatly acknowledged. This work is supported by National Natural Science Foundations of China (No. 31301965), Anhui Provincial Natural Science Foundations (No. 1308085QC63, No. 1408085QC65) and Anhui Provincial Educational Commission Natural Science Foundations (No. KJ2012A216, No. KJ2013B198). This work is also supported by Fuyang Normal College Committee’s Educational Project (No. 2012JYXM71) and the 2014 program for Anhui provincial outstanding young talents in colleges and universities.
REFERENCES
- Aranyi, T., A. Varadi, I. Simon and G.E. Tusnady, 2006. The BiSearch web server. BMC Bioinform., Vol. 7.
CrossRefDirect Link - Bekaert, M. and E.C. Teeling, 2008. UniPrime: A workflow-based platform for improved universal primer design. Nucl. Acids Res., Vol. 36.
CrossRefDirect Link - Bellemain, E., T. Carlsen, C. Brochmann, E. Coissac, P. Taberlet and H. Kauserud, 2010. ITS as an environmental DNA barcode for fungi: An in silico approach reveals potential PCR biases. BMC Microbiol., Vol. 10.
CrossRefDirect Link - Boutros, P.C. and A.B. Okey, 2004. PUNS: Transcriptomic-and genomic-in Silico PCR for enhanced primer design. Bioinformatics, 20: 2399-2400.
CrossRefPubMedDirect Link - Cao, Y., L. Wang, K. Xu, C. Kou and Y. Zhang et al., 2005. Information theory-based algorithm for in silico prediction of PCR products with whole genomic sequences as templates. BMC Bioinform., Vol. 6.
CrossRefDirect Link - Ficetola, G.F., E. Coissac, S. Zundel, T. Riaz and W. Shehzad et al., 2010. An in silico approach for the evaluation of DNA barcodes. BMC Genomics, Vol. 11.
CrossRefDirect Link - Fredslund, J. and M. Lange, 2007. Primique: Automatic design of specific PCR primers for each sequence in a family. BMC Bioinform., Vol. 8.
CrossRefDirect Link - Gadberry, M.D., S.T. Malcomber, A.N. Doust and E.A. Kellogg, 2005. Primaclade: A flexible tool to find conserved PCR primers across multiple species. Bioinformatics, 21: 1263-1264.
CrossRefPubMedDirect Link - Housley, D.J.E., Z.A. Zalewski, S.E. Beckett and P.J. Venta, 2006. Design factors that influence PCR amplification success of cross-species primers among 1147 mammalian primer pairs. BMC Genom., Vol. 7.
CrossRefDirect Link - Karolchik, D., R. Baertsch, M. Diekhans, T.S. Furey and A. Hinrichs et al., 2003. The UCSC genome browser database. Nucleic Acids Res., 31: 51-54.
CrossRefDirect Link - Kalendar, R., D. Lee and A.H. Schulman, 2011. Java web tools for PCR, in silico PCR and oligonucleotide assembly and analysis. Genomics, 98: 137-144.
CrossRefDirect Link - Karolchik, D., G.P. Barber, J. Casper, H. Clawson and M.S. Cline et al., 2014. The UCSC genome browser database: 2014 update. Nucleic Acids Res., 42: D764-D770.
CrossRef - Kent, W.J., C.W. Sugnet, T.S. Furey, K.M. Roskin, T.H. Pringle, A.M. Zahler and D. Haussler, 2002. The human genome browser at UCSC. Genome Res., 12: 996-1006.
CrossRefPubMedDirect Link - Lexa, M. and G. Valle, 2003. PRIMEX: Rapid identification of oligonucleotide matches in whole genomes. Bioinformatics, 19: 2486-2488.
CrossRefPubMedDirect Link - Liu, W.Y., C.J. Zhao and J.Y. Li, 2010. A non-invasive and inexpensive PCR-based procedure for rapid sex diagnosis of Chinese gamecock chicks and embryos. J. Anim. Vet. Adv., 9: 962-970.
CrossRefDirect Link - Marshall, O.J., 2004. PerlPrimer: Cross-platform, graphical primer design for standard, bisulphite and real-time PCR. Bioinformatics, 20: 2471-2472.
CrossRefPubMedDirect Link - Mullis, K., F. Faloona, S. Scharf, R. Saiki, G. Horn and H. Erlich, 1986. Specific enzymatic amplification of DNA in vitro: The polymerase chain reaction. Cold Spring Harb. Symp. Quant. Biol., 51: 263-273.
CrossRefPubMedDirect Link - Mullis, K., F. Faloona, S. Scharf, R. Saiki, G. Horn and H. Erlich, 1992. Specific enzymatic amplification of DNA in vitro: The polymerase chain reaction. 1986. Biotechnology, 24: 17-27.
PubMed - Nishigaki, K., A. Saito, H. Takashi and M. Naimuddin, 2000. Whole genome sequence-enabled prediction of sequences performed for random PCR products of Escherichia coli. Nucleic Acids Res., 28: 1879-1884.
Direct Link - Noda, A.A., I. Rodriguez, B. Mondeja and C. Fernandeze, 2013. Design, optimization and evaluation of a polymerase chain reaction for detection of Borrelia spp. Adv. Clin. Exp. Med., 22: 639-653.
PubMedDirect Link - Rubin, E. and A.A. Levy, 1996. A mathematical model and a computerized simulation of PCR using complex templates. Nucleic Acids Res., 24: 3538-3545.
CrossRefPubMedDirect Link - Saiki, R.K., S. Scharf, F. Faloona, K.B. Mullis, G.T. Horn, H.A. Erlich and N. Arnheim, 1985. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science, 230: 1350-1354.
CrossRefDirect Link - Saiki, R., D.H. Gelfand, S. Stoffel, S.J. Scharf and R. Higuchi et al., 1988. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science, 239: 487-491.
CrossRefPubMedDirect Link - Yuryev, A., J. Huang, M. Pohl, R. Patch and F. Watson et al., 2002. Predicting the success of primer extension genotyping assays using statistical modeling. Nucleic Acids Res., Vol. 30, No. 23.
CrossRefDirect Link - Zhao, C.J., C.F. Wang, X.M. Deng, Y. Gao and C.H. Wu, 2006. Identification of single-nucleotide polymorphisms in 5' end and exons of the PRKAG3 gene in Hubbard White broiler, Leghorn layer and three Chinese indigenous chicken breeds. J. Anim. Breed. Genet., 123: 349-352.
CrossRefPubMedDirect Link