
M. J. Harighi
Faculty of Science, Razi University, Iran
M. R. Zamani
National Institute of genetic Engineering and Biotechnology, Tehran, Iran
M. Motallebi
National Institute of genetic Engineering and Biotechnology, Tehran, Iran







ABSTRACT
Chitin, a linear polymer of N-acetylglucosamine residues, has been the most abundant polymer in nature after cellulose. It plays a major role in fungal cell walls. Chitinase enzymes degrade the polymer into its component residues by breaking the β-1,4 glycosidic bonds. As a producer of a variety of chitinase enzymes, the filamentous fungus, Trichoderma, has become an important means of biological control for fungal diseases. Chitinase 42 kDa (Chit42) was purified from supernatants of T. atroviride, an over producer of chitinase enzyme, grown in medium supplemented with colloidal chitin as the sole carbon source. Enzyme purification was achived in two steps ion exchange chromatography using CM-Sepharose and DEAE- Sepharose. The purity of Chit42 was investigated by SDS-PAGE. The results showed one band about 42 kDa after the second step of chromatography which showed chitinase activity in enzyme assay technique. This purified Chit42 has shown to have inhibitory activity on mycelial growth and also, in vitro lytic activity on cell wall of Rhizoctonia solani (AG2-2), causal agent of root rot in sugar beet. The purified Chit42 was optimally active at pH 5 and at 40°C. It is thermally stable at 60°C for more than 240 min at pH 5.
PDF Abstract XML References
How to cite this article
M. J. Harighi, M. R. Zamani and M. Motallebi, 2007. Evaluation of Antifungal Activity of Purified Chitinase 42 from Trichoderma atroviride PTCC5220. Biotechnology, 6: 28-33.
DOI: 10.3923/biotech.2007.28.33
URL: https://scialert.net/abstract/?doi=biotech.2007.28.33
DOI: 10.3923/biotech.2007.28.33
URL: https://scialert.net/abstract/?doi=biotech.2007.28.33
INTRODUCTION
Chitin, a linear β-(1,4)-linked N-acetylglucosamine (GlcNAc) polysaccharide is a main structure component of fungal cell wall (Cabib, 1991; Gooday, 1990). Chitinases (EC 3.2.1.14), essential enzymes catalyzing the conversion of chitin to its monomeric or oligomeric components, have been found in a wide range of organisms, including bacteria (Cody et al., 1990; Gooday, 1990; Perrakis et al., 1994), plants (Shinshi et al., 1987; Gijzen et al., 2001; Masuda et al., 2001) and fungi (Bratnicki-Gracia, 1969; Omumasaba et al., 2001). The production of chitinase by plants has been suggested to be a part of their defense mechanism against fungal pathogens (Gijzen et al., 2001; Masuda et al., 2001). Those produced by bacteria appear to have a nutritional or scavenging role, as shown by their secretion into the medium (O’Brien and Colwell, 1987). In fungi, chitinase activity probably has a physiological role in hyphal growth and morphognesis (Bartnick-Garcia, 1973; Wessels, 1986; Gooday et al., 1986). Several workers have suggested that chitinase-producing fungi, such as Trichoderma sp., can be effective as biocontrol of soil borne plant pathogenic fungi (Chet, 1987). Production of chitinase by a number of Trichoderma strains has been reported, when it is grown in the presence of chitin or isolated fungal cell wall (Elad et al., 1983; Ridout et al., 1988). This study describes the purification and some properties of an extracellular chitinase (Chit42) produced by T. atroviride.
MATERIALS AND METHODS
Microorganisms and cultural conditions: Trichoderma atroviride PTCC5220 obtained from Persian Type Culture Collection. It was identified in our laboratory among 30 Trichoderma isolates as a high producer of chitinolytic enzymes. The stock culture was stored on agar (1.5%) slant of MY medium (2% malt extract, 0.2% yeast extract, 1% maltose). Rhizoctonia solani (AG 2-2), causal agent of root rot in sugar beet, was kindly supplied by Prof. Banihashemi, Mycology Lab., Department of Plant Pathology, College of Agriculture, Shiraz University, Shiraz, I.R. of Iran. The fungus was propagated on Potato Dextrose Agar (PDA) and subcultured as needed.
Enzyme production: For enzyme production, T. atroviride was grown in 200 mL of Czapeck-Dox medium containing the following per litter, 3 g NaNO3, 0.5 g MgSO4.7H2O, 0.5 g KCl, 0.01 g FeSO4.7H2O, 1 g KH2PO4 and supplemented with 10% glucose in 500 mL flask. The flask was inoculated with 2 mL conidial suspension (106 conidia/mL) of T. atroviride and incubated for 96 h at 25°C as stationary culture. Harvested mycelia were washed several times with 2% of MgCl2 and distilled water and transferred to Czapeck-Dox medium supplemented with 1.5% colloidal chitin. The culture medium was incubated at 25°C at 100 rpm, harvested after 3 days, filtered, and centrifuged. The supernatant was used for further purification. Protein concentration was estimated spectrophotometrically at 280 nm by the method of Bradford (1976) using bovine serum albumin as standard protein.
Enzyme purification: The crude chitinase was put on a CM-Sepharose Fast Flow (Pharmacia) cation exchange column (2x25 cm) equilibrated with 50 mM sodium acetate buffer pH 4. After washing out the unabsorbed protein with the same buffer, the adsorbed protein was eluted with 0.0- 0.5 M NaCl linear gradient in the same buffer. Fractions of 5 mL were collected and assayed for chitinase activity.
The fractions with chitinase activity were pooled and put on a DEAE- Sepharose Fast Flow (Pharmacia) anion exchange column (1.5x16 cm) equilibrated with 50 mM sodium acetate buffer pH 7.5. The column was washed at a flow rate of 1.5 mL min-1 with the same buffer until the absorbance (A280) returned to near the base line. The bond proteins were then eluted with a 0.0- 0.5 M NaCl linear gradient in the same buffer. Fractions of 5 mL were collected and appropriate fractions were pooled and stored at -80°C.
Temperature optimum and stability: The temperature optimum was determined by performing the standard assay within the temperature range of 10 to 100°C. The inactivation temperature was also determined by incubation the enzyme for 300 min at temperature from 30 to 80°C in 15 mM sodium acetate buffer, pH 5 and then measuring the remaining activity at 37°C adding colloidal chitin as the assay substrate. The inactivation temperature was defined as the temperature at which the activity was reduced by 50%, under the same conditions.
pH optimum: The optimum pH of purified enzyme was determined after incubation with colloidal chitin in the buffer at various pH (1 to 10). The buffers used were: 10 mM HCl (pH 1), 15 mM phosphate buffer (pH 2), 15 mM citrate buffer (pH 3), 15 mM acetate buffer (pH 4-5), 15 mM MES (Morpholino Ethan Sulfunic acid) (pH 6), 15 mM phosphate buffer (pH 7), 15 mm Tris-HCl buffer(pH 8), and 15 mM CHES (Cyclo Hexylamino Ethan Sulfunic acid) (pH 9,10).
SDS-polyacrylamide gel electrophoresis: SDS-PAGE was done by the method of Lammeli (1970) using a 12.5% acrylamide gel. Proteins on the gel were stained with coomassie brilliant blue R-250.
Preparation of colloidal chitin: Colloidal chitin was prepared from purified chitin according to the method of Roberts and Selitrennikott (1998) with minor modification. Ten grams of chitin powder were added slowly into 100 mL of H3PO4 (85%) at 25°C under vigorous stirring for 2 h. The suspension was poured into 1 L of ice-cold 95% alcohol under vigorous stirring for 30 min and stored at -20°C until use. When in need, 10 mL of the suspension was centrifuged. The precipitate was washed with 50 mL of 0.1 mol sodium phosphate buffer (pH 7.0) for 3 times. The derived precipitate was dissolved in 90 mL of 0.1 mol sodium phosphate buffer (pH 6.0), which was about 10 g L-1 colloidal chitin solution.
Enzyme assay: Chitinase activity was assayed with 200 μL -1 of colloidal chitin (5 mg mL-1), and 200 μL of purified enzyme. The mixture was incubated for 60 min at 40°C and the reaction was stopped by adding 1 mL of NaCl (1%) and centrifuged at 6000xg for 5 min. The supernatant was boiled with 100 μL of potassium tetra borate buffer for 3 min. Three milliliter of DMAB reagent [10 g of dimethyl amino benzaldehyde in 100 mL of glacial acetic acid (12.5%) and 10 N chloridric acid (87.5%)] was added to the reaction and incubated at 40°C for 20 min and the amount of N-acetylglucosamine (GLcNAc) produced in the supernatant was determined by the method described by Zeilinger et al. (1999) using GLcNAc as standard. One unit of enzyme activity was defined as the amount of enzyme that catalyzes release of 1 μmol GLcNAc in 60 min at 40°C.
Antifungal activity: The antifungal activity of purified Chit42 was tested by using a modification of the bioassay described by Broglie et al (1991). An agar disk (5 mm in diameter) with the fungus R. solani, which was derived from the fungus in an actively growing state previously cultured on PDA, was placed in the center of a Petri dish containing PDA. The plates were incubated at 25°C for 2 days. Wells were subsequently punched into the agar at a distance of 25 mm from the center of the plates. The samples to be tested were placed into the wells in 40 μL-1 of sterile dH2O. The plates were incubated for 72 h at 34 °C and then photographed.
Light microscopy: To study the effect of purified Chit42 on mycelium cell wall of R. solani. Sixty units of purified Chit42 was added to 2 days grown of R. solani on a slide covered with a thin layer of PDA. The slide was examined for degradation of R. solani cell wall under a light microscope.
RESULTS
Production and purification of chit42: To produce extracellular chitinase from T. atroviride PTCC5220, a high producer isolate of chitinolytic enzyme, the mycelia from this fungus previously grown for 96 h in glucose-supplemented Czapeck-Dox medium was inoculated into Czapeck-Dox medium supplemented with colloidal chitin and the secreted enzymes into the medium were used for purification. Filtrate culture was removed by centrifugation, and the proteins were concentrated by ammonium sulfate precipitation. The recovered proteins were then fractionated by two steps ion-exchange chromatography, using CM-Sepharose and DEAE-Sepharose. All procedures were done in cold room. A fraction peak containing chitinase activity was obtained through CM-Sepharose column chromatography (not shown). Chitinase 42 (Chit42), the main enzyme, was purified by DEAE- sepharose column (Fig. 1). Fractions 36-40 in the second absorbance (A280) peak, which was eluted at approximately 0.25-0.35 M NaCl, contained chitinase activity. Extracellular Chit42 was purified up to 61.8 fold from culture supernatant. Analysis of the pooled fractions (36-40) with chitinase activity by SDS-PAGE revealed the presence of a single protein band along with two other weak bands (Fig. 2), while fractions 37 and 38 with highest chitinase activity (1 U mg-1) showed to contain Chit42 as a single sharp band with no trace contaminants (Fig. 2), suggesting that the enzyme is monomer. The molecular weight of the purified enzyme was calculated to be about 42 kDa.
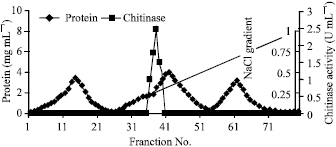 | |
| Fig. 1: | Ion-exchange chromatography of T. atroviride chitinase enzyme on DEAE-Sepharose Fast Flow. The column was washed with 50 mM sodium acetate buffer, pH 7.5 and adsorbed proteins were eluted with a linear gradient of NaCl in the same buffer. Eighty fractions of 5 mL were collected |
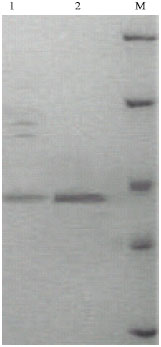 | |
| Fig. 2: | SDS-PAGE of purified Chit42: line 1) pooled fractions 36-40, line 2) fractions 37 and 38, M) molecular marker |
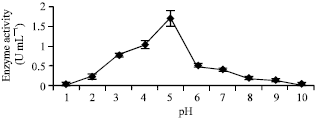 | |
| Fig. 3: | Effect of pH on enzymatic activity of the purified Chit42 |
Effect of pH and temperature on chitinase activity: The effect of pH on the enzyme activity of purified Chit42 was determined by varying the pH of the reaction mixture (pH 1 to 10) using different buffers. The denaturing effects of pH on chitinase protein were investigated by incubating the enzyme solution for 60 min at various pH values at 40°C. The solutions were then adjusted to pH 5 and residual chitinase activity was measured by standard assay. The maximal enzyme activity of purified Chit42, was revealed at pH 5 (Fig. 3).
The effects of temperature on the enzyme activity of purified Chit42 were examined at pH 5. Chitinase was assayed under standard conditions, except for temperatures which were varied from 30-80°C. The optimum temperature for purified Chit42 activity was found to be 40°C (Fig. 4).
The stability of the purified enzyme was examined by maintaining the enzyme solution at different temperatures (30 to 80°C with 10°C intervals) for 300 min, followed by activity assay under standard conditions. Not significant decrease in enzyme activity was observed at 30 to 60°C after incubation for 240 min while the activity of purified Chit42 decreased by 50% after incubation for 130 min at 70°C and 100 min at 80°C (Fig. 5).
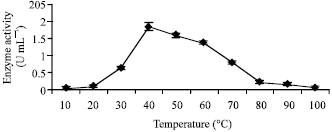 | |
| Fig. 4: | Effect of temperature on enzymatic activity of the purified Chit42 |
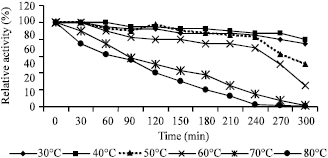 | |
| Fig. 5: | Effect of different temperatures on stability of the purified Chit42. The remained enzymatic activities after incubation at different temperatures were measured |
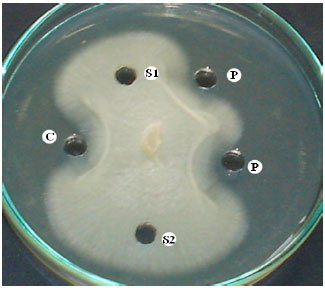 | |
| Fig. 6: | Inhibition of fungal growth by Chit42: C) crude enzyme obtained from culture medium of T. atroviride, P) Purified Chit42 from culture medium of T. atroviride, S1) culture medium without inoculation (negative control), and S2) sodium acetate buffer without enzyme (negative control) |
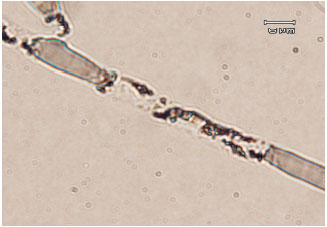 | |
| Fig. 7: | A light micrograph showing degradation of R. solani cell walls by purified Chit42 |
Antifungal activity: For evaluation of antifungal activity of purified Chit42 on actively growing phytopathogenic fungus R. solani mycelia, 40 μL-1 of 50 mM sodium acetate buffer pH 5 containing 60 units of purified enzyme was added to the punched wells in PDA medium. The growth of R. solani was inhibited by addition of the purified enzyme as compared to 50 mM sodium acetate buffer without chitinase enzyme (Fig. 6). Similar observation was obtained when the same amount of crude enzyme (60 units) from T. atroviride culture medium was added to growth culture of R. solani (Fig. 6).
Also, the effect of purified enzyme on cell wall of R. solani was tested microscopically by adding 20 μL-1 of 50 mM sodium acetate buffer pH 5 containing 60 units of purified Chit42 on 2 days grown of R. solani mycelia on a slide covered with PDA. After 60 min of incubation at 40°C, the effect of enzyme was evaluated and the results showed that the purified Chit42 are able to lyse the R. solani mycelium cell wall (Fig. 7). No lysis activity was observed when 50 mM sodium acetate buffer without chitinase enzyme was used as a control.
DISCUSSION
The direct mycoparasitic activity of Trichoderma species has been reported to be the major mechanism proposed to explain their antagonistic activity against phytopathogenic fungi, their lytic activity being mainly due to the chitinase and glucanse hydrolases (De la Cruz et al., 1992). The substrate of chitinase is chitin, which is a common component of fungal cell walls (Broglie et al., 1991). These enzymes are strong inhibitors of many important plant pathogens and the chitinases are able to lyse the hard chitin cell wall of the mature hyphae, conidia, chlamydospores and sclerotia (Lorito et al., 1998).
In the past two decades, extensive studies on chitinases have been done by a large number of laboratories. This high level of interest in chitinases is mostly due to the antifungal property of these enzymes. Most of these studies were on the characterization of the genes and cDNA and on examination of gene expression and its regulation. On the other hand, only a few studies actually involved purified enzymes. This reflects, at least to some degree, experimental difficulties generally associated with protein purification work.
Trichoderma chitinases are substantially more antifungal than any other chitinases purified from any other sources when assayed under the same conditions. They are more active than corresponding plant enzymes, effective on a much wider range of pathogens, and are nontoxic to plants at high concentrations (Wang et al., 2003; Lorito et al., 1998).
In this study a highly producer of chitinolytic enzymes Iranian isolate of T. atroviride was used for purification of Chit42. T. atroviride Chit42 was purified about 62 folds from the culture filtrate. Ammonium sulfate precipitation has been used by other workers as the first step of purification of chitinases from different organisms (Ulhoa and Peberdy, 1992). This method was able to recover about 52 % of enzyme activity of T. atroviride chitinase and contaminants were eliminated. The procedure that we used to purify further Chit42 was two steps purification using CM-Sepharose and DEAE-Sepharose chromatography. Most of the contaminant proteins were removed by ion exchange chromatography on CM-Sepharose. The enzyme recovered was further purified by chromatography on a DEAE-Sepharose column. SDS-PAGE analysis of the purified enzyme showed the homogeneity of this chitinase and revealed one protein band with an estimated molecular weight of 42 kDa with no trace contaminants. The molecular weights of endochitinases from different strains of Trichoderma harzianum are varied from 31 to 52 kDa (Haran et al., 1996; De La Cruz et al., 1992; Haraman et al., 1993). However, the molecular weight of the purified chitinase enzyme from T. harzianum strain 39.1 was 40 kDa (Ulhoa and Peberdy, 1992). The purified Chit42 enzyme from T. atroviride is similar to that of Aspergillus nidulans and Choanephora cucurbitarum in those they have a low pH optimum for enzyme activity. The optimum pH for A. nidulans was 5, for C. cucurbitarum was between 4.5-6 and for many other fungal chitinases are around 4.0-8.0 (Sahai and Manocha, 1993).
The purified Chit42 effectively hydrolyses colloidal chitin at pH 5 and the half-life of the purified protein at 60°C (4 h) or 70°C for 2 h suggests that this purified enzyme is one of the heat-stable chitinases. Also, the lytic activity of purified Chit42 was evaluated in vitro on phytopathogenic fungus R. solani. Effective growth inhibitory was observed when purified Chit42 was added to R. solani on solid medium. The observed growth inhibitory effect may arise from enzyme catalyzed hydrolysis of newly formed chitin and resultant disruption of the growing fungal mycelium (Broglie et al., 1991).
Microscopic observation demonstrated the ability of purified Chit42 to degrade cell walls of R. solani. Similar to our findings, a number of other studies have implicated chitinase enzyme to be responsible for the degradation of phytopathogenic fungi cell wall (Elad et al., 1982; Ridout et al., 1988; Tweddell et al., 1994).
Information obtained from such studies would be useful in understanding the effect of chitinase against phytopathogenic fungi.
REFERENCES
- Bratnicki-Gracia, S., 1969. Cell wall chemistry, morphogenesis and taxonomy of fungi. Annu. Rev. Microbiol., 22: 87-108.
CrossRef - Bradford, M.M., 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem., 72: 248-254.
CrossRefPubMedDirect Link - Brogue, K., I. Chet, M. Holliday, R. Cressman and P. Biddle et al., 1991. Transgenic plants with enhanced resistance to the fungal pathogen Rhizoctonia solani. Science, 254: 1194-1197.
CrossRefDirect Link - Cody, R.M., N.D. Davis, J. Lin and D. Shaw, 1990. Screening microorganisms for chitin hydrolysis and production of ethanol from amino sugars. Biomass, 21: 285-295.
CrossRefDirect Link - De La Cruz, J., A. Hidalgo-Gallego, J.M. Lora, T. Benitez, J.A. Pintor-Taro and A. Llobell, 1992. Isolation and characterization of three chitinase in Trichoderma harzianum. Eur. J. Biochem., 206: 859-867.
CrossRef - Elad, Y., I. Chet and Y. Henis, 1982. Degradation of plant pathogenic fungi by Trichoderma harzianum. Can. J. Microbiol., 28: 719-725.
CrossRefDirect Link - Elad, Y., I. Chet, P. Boyle and Y. Henis, 1983. Parasitism of Trichoderma spp. on Rhizoctonia solani and Sclerotium rolfsii-Scanning electron microscopy and fluorescence microscopy. Phytopathology, 73: 85-88.
CrossRefDirect Link - Gijzen, M., K. Kuflu, D. Qutob and J.T. Chernys, 2001. A class I chitinase from soybean seed coat. J. Exp. Bot., 52: 2283-2289.
Direct Link - Gooday, G.W., A.M. Humphreys and W.H. McIntosh, 1986. Roles of Chitinases in Fungal Growth. In: Chitin in Nature and Technology, Muzzarelli, R.A.A., C. Jeuniaux and G.W. Gooday (Eds.), Plenum Press, New York (USA)., pp: 83-91.
Direct Link - Haran, S., H. Schickler and I. Chet, 1996. Molecular mechanisms of lytic enzymes involved in the biocontrol activity of Trichoderma harzianum. Microbiology, 142: 2321-2331.
Direct Link - Laemmli, U.K., 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227: 680-685.
CrossRefDirect Link - Lorito, M., S.L. Woo, I. Garcia, G. Colucci and G.E. Harman et al., 1998. Genes from mycoparasitic fungus as a source for improving plant resistance to fungal pathogens. Proc. Natl. Acad. Sci. USA., 95: 7860-7865.
PubMed - Masuda, S., H. Kamada and S. Satoh, 2001. Chitinase in cucumber xylem sap. Biosci. Biotechnol. Biochem., 65: 1883-1885.
Direct Link - Omumasaba, C.A., N. Yoshida and K. Ogawa, 2001. Purification and characterization of a chitinase from Trichoderma viride. J. General Applied Microbiol., 47: 53-61.
Direct Link - Perrakis, A., I. Tews, Z. Dauter, A.B. Oppenheim, I. Chet, K.S. Wilson and C.E. Vorgias, 1994. Crystal structure of a bacterial chitinase at 2.3 A resolution. Structure, 2: 1169-1180.
CrossRef - Ridout, C.J., J.R. Coley-Smith and J.M. Lynch, 1988. Fractionation of extracellular enzymes from a mycoparasitic strain of Trichoderma harzianum. Enzyme Microb. Technol., 10: 180-187.
CrossRefDirect Link - Roberts, W.K. and C.P. Selitrennikoff, 1998. Plant and bacterial chitinases differ in antifungal activity. J. Gen. Microbiol., 134: 169-176.
CrossRefDirect Link - Sahai, A.S. and S.M. Manocha, 1993. Chitinases of fungi and plants: Their involvement in morphogenesis and host-parasite interaction. FEMS Microbiol. Rev., 11: 317-338.
CrossRefDirect Link - Shinshi, H., D. Mohnen and F. Meins, 1987. Regulation of a plant pathogenesis-related enzyme: Inhibition of chitinase and chitinase mRNA accumulation in cultured tobacco tissues by auxin and cytokinin. Proc. Natl. Acad. Sci. USA., 84: 89-93.
Direct Link - Tweddell, R.J., S.H. Jabaji-Hare and P.M. Charest, 1994. Production of chitinases and β-1,3 glucanases by Stachybotrys elegans, a mycoparasitie of R. solani. Applied Environ. Microbiol., 60: 489-495.
Direct Link - Ulhoa, C.J. and J.F. Peberdy, 1992. Purification and some properties of the extracellular chitinase produced by Trichoderma harzianum. Enzyme Microb. Technol., 14: 236-240.
CrossRef - Wang, Y., A.P. Kausch, J.M. Chandlee, H. Luo and B.A. Ruemmele et al., 2003. Co-transfer and expression of chitinase, glucanase and bar genes in creeping bentgrass for conferring fungal disease resistance. Plant Sci., 165: 497-506.
Direct Link - Wessels, J.G.H., 1986. Cell wall synthesis in apical hyphal growth. Int. Rev. Cytol., 104: 37-39.
Direct Link - Zeilinger, S., C. Galhaup, K. Payer, S.L. Woo and R.L. Mach et al., 1999. Chitinase gene expression during mycoparasitic interaction of Trichoderma harzianum with its host. Fungal Genet. Biol., 26: 131-140.
CrossRefDirect Link
sathiyamoorthy Reply
excellent work and interesting work most of very use for my work guideline.